

-
使用NH3的选择性催化还原 (NH3-SCR) 已被广泛用于去除固定污染源烟气中的氮氧化物[1]。基于VO5-WO3/TiO2的商用SCR催化剂最常被用于NH3-SCR系统。然而,由于安装空间有限,高工作温度 (>300 ℃) 限制了这种催化剂在工业窑炉系统中的使用。因此,低温SCR催化剂已被广泛研究。在这类催化剂中,氧化铈具有高储氧能力 (oxygen storage capacity) ,且Ce4+与Ce3+之间能实现可逆转换[2],因此是一种重要的脱硝催化剂活性组分。近年来,已有多种低温NH3-SCR的氧化铈基催化剂被开发,包括CeO2/TiO2基[3]、CeO2/Al2O3基[4-5]、CeO2-MnO2 [6-7]。这些催化剂均可在中低温 (<300 ℃) 下表现出较高的脱硝活性和N2选择性。
烟气中SO2组分的存在会在短时间内对低温脱硝活性造成不可逆转的毒害作用,使其失活。许多研究者对SO2对Ce/TiO2催化剂在中低温下的中毒机理进行了深入研究。ZHANG等[8]发现CeO2的硫酸化减少了Ce-O-Ti活性位数量,进一步导致催化剂在300 ℃下活性降低;XU等[9]使用DRIFT、SO2-TPD等表征技术对Ce/TiO2在300 ℃下的中毒机理进行研究,发现前12 h内活性下降是由于硫酸铵盐的沉积导致,后36 h是由于Ce(SO4)2和Ce2(SO4)3的生成;DONG等[10]在250 ℃下对V/Sb/TiO2和V/Ce/Sb/TiO2催化剂进行了的抗硫中毒研究,发现Ce2(SO4)3的生成减少了表面硫酸铵盐的沉积,故其抗硫性能得以提升;XU等[11]发现在200 ℃下CeO2-WO3/TiO2催化剂的失活是由于硫酸铵盐和硫酸铈的沉积共同导致。因此,硫酸铵盐和硫酸铈的沉积是铈基催化剂在含硫SCR气氛下失活的主要原因。少量研究者对不同温度下的SO2中毒进行了研究,HUANG等[12]研究了在SO2和H2O共同存在的气氛下,不同反应温度下Mn-Fe/MPS催化剂的失活情况,发现在170 ℃时脱硝活性比190 ℃时下降得更快,但详细的机制还未被揭示。MA等[13]研究了Fe-Cu /CNTs-TiO2催化剂在不同温度下的失活情况,发现在150、200和250 ℃下不同的活性演变可归结为(NH4)2SO4的沉积,并降低了NH3的吸附、NO的吸附和氧化能力。 XU等[14]也在300和350 ℃下进行了SO2气氛下的NH3-SCR实验,结果表明较低的反应温度增强了SO2对脱硝活性的负面影响,但是其机理并未被进一步揭示。
为探究Ce-TiO2催化剂在不同温度下的不同中毒机制和反应机理,采用溶胶-凝胶法制备了Ce-TiO2 (Ce/Ti摩尔比为0.25) ,并在SO2存在的气氛下对催化剂进行不同温度 (180 ℃、240 ℃) 下的抗硫中毒测试,结合不同时间下的中毒活性演变、硫组分定量测试及一系列的表征测试,得出催化剂在不同温度下的不同中毒机制以及反应机理,以期为了解SO2中毒效应与反应温度之间的内在联系,以及Ce-TiO2催化剂在SO2气氛下的失活机理及其活性改善提供参考。
-
采用溶胶凝胶法制备Ce-TiO2及Ce-Co-TiO2催化剂,具体流程如下:取适量硝酸铈、硝酸钴、无水乙醇、硝酸、去离子水倒入烧杯,配置成A液并充分搅拌;取适量钛酸四丁酯、无水乙醇、冰醋酸倒入烧杯,配置成B液并充分搅拌,随后将A液缓缓滴入B液并持续搅拌,滴定完成后继续搅拌3 h;随后室温静置12 h后形成透明凝胶,之后置于80 ℃的烘箱中干燥24 h直至形成固体粉末;最后置于450 ℃的马弗炉煅烧6 h,研磨至40~80目,装样密封保存。
-
催化剂的活性评价与抗SO2中毒实验都在内径为18 mm的固定床反应管内进行。反应温度为150~330 ℃,称取0.2 g催化剂粉末并置于反应管内床层上方。本实验模拟烟气组成为:1 340 mg∙L−1 NO、760 mg∙L−1 NH3、570 mg∙L−1 SO2 (抗硫测试) 、5% O2、N2作为平衡气、气体总流量为700 mL∙min−1 ,空速为42 000 h−1,各路气体首先经过预混器并进行充分混合,然后进入反应管内进行反应。经过反应后的NOx、N2O通过Testo350烟气分析仪、BedfontG200分析仪进行测试,催化剂的脱硝活性、N2O选择性由公式 (1) 和 (2) 计算。
式中:η表示脱硝活性,S表示N2O选择性。[NOx]out和[NOx]in分别代表反应器出口、入口测得的NOx质量浓度,[N2O]out代表反应器出口所测得的N2O质量浓度,[NH3]in和[NH3]out分别代表入口、出口的NH3质量浓度。Fresh-Ce-TiO2表示新鲜未中毒的Ce-TiO2催化剂;P-180-Ce-TiO2表示在180 ℃下SO2氛围中进行 NH3-SCR反应2 h后的V/TiO2催化剂;P-240-Ce-TiO2表示在240 ℃下SO2氛围中进行NH3-SCR反应8 h后的Ce-TiO2催化剂。
-
BET测试使用JW-BK112仪器在-196 ℃的整个相对压力范围内获得N2的吸附-解吸等温线,并通过BET方程和BJH方程从吸附/解吸等温线计算出比面积、孔体积和平均孔径。NH3-TPD测试使用彼奥德化学吸附分析仪:在常温下通入1%NH3/N2吸附1 h,再进行1 h的He吹扫,最后在He流中以10 ℃·min−1的升温速率将温度提高到900 ℃,利用热导检测器监测从催化剂上解吸的NH3。H2-TPR分析仍使用彼奥德化学吸附分析仪进行:通入10%H2/Ar气流并以10 ℃·min−1升温速率升至900 ℃,通过检测器检测H2消耗量。XPS测试使用ESCALAB Mark II光谱仪,用Al KR辐射(1486.6 eV)观察表面成分的含量和化学状态,使用被污染的碳 (BE=284.6 eV) 对结合能进行了校正。XRD测试使用Philips X pert Pro衍射仪,扫描速度为5(°)·min−1;TG-DTG测试使用Netzsch热分析仪STA449C,50 mL·min−1氮气氛围下,升温速率为10 ℃·min−1。ICP测试采用 NexION1000G仪器进行测试:首先称取50 mg中毒后样品置于200 mL离心管中,加入一定量去离子水,经过1 h超声、过滤、定容后进行ICP测试。原位漫反射红外傅里叶变换光谱 (in-situ DRIFTS) 测试是在NicoletNexus 5700 FTIR光谱仪上进行的,该光谱仪配备了1个Harrick IR池和1个MCT检测器。
-
图1为Ce-TiO2催化剂及不同含量的Co掺杂改性后催化剂的活性和抗硫性。Ce-TiO2催化剂在150 ℃下脱硝活性仅30.8%,随着反应温度升高至210 ℃,脱硝活性上升至100%,进一步升高至330 ℃,脱硝活性反而下降至89.7%。这可能是由于高温下NH3发生了过度氧化,进一步产生NO,降低了脱硝反应活性[15]。当Co掺杂比例为Co/Ti为0.1时,150 ℃下脱硝活性升高至75%,低温活性大大增加。随着温度升高至300 ℃,脱硝活性下降至79.4%。分析在不同反应温度下,SO2对Ce-TiO2和Ce-Co-0.1-TiO2催化剂的活性和N2O选择性影响:在180 ℃下,未通入SO2时,Ce-TiO2催化剂的脱硝效率达到50.7%,N2O选择性为2.5%,通入1 h质量浓度为570 mg∙L−1的SO2后,NO转化率下降至18.4%,并在接下来的1 h内缓慢降低至12.5%,N2O选择性缓慢降低至0.2%,且切断SO2后活性无明显变化。在Co掺杂后,1 h内活性下降至44.1%,并在接下来的1 h内降至24.8%,N2O选择性缓慢降至1.4%。在240 ℃下,SO2通入8 h后,Ce-TiO2催化剂活性缓慢降至30.9%,N2O选择性从8.1%降至2.0%,Co掺杂后催化剂活性下降至44.2%,N2O选择性从13.4%降低至4.9%,SO2切断后脱硝活性无明显变化。以上结果表明,SO2与NO的在催化剂表面的竞争吸附不是活性降低的主要原因,催化剂物化特性的改变导致了催化剂活性的永久丧失。同时,对比不同温度下的活性变化发现,反应温度越低,催化剂活性下降越迅速。这表明SO2对催化剂脱硝活性的中毒作用与反应温度有着密切联系。为更深入地研究反应温度与SO2中毒之间的关系,对不同温度中毒后的催化剂进行表征。
-
1) XRD和BET分析。XRD和BET结果见图2。新鲜Ce-TiO2催化剂含有锐钛型TiO2、CeO2晶相,经过180 ℃和240 ℃中毒后催化剂的晶相并未发生变化,无新的含硫晶相产生。Co改性后催化剂晶相并未发生变化,Co3O4晶相并未发现。这可能是由于Co3O4较为均匀分布在TiO2载体上,无法被X射线检测到,且中毒后催化剂晶相无明显变化。新鲜Ce-TiO2催化剂的最可几孔径为9.6 nm,中毒后孔径分布无明显变化;Co掺杂后催化剂的最可几孔径为11.4 nm,且中毒后无明显变化。表1为催化剂的结构参数。Co掺杂后催化剂比表面积从120.9 m2·g−1降至98.7 m2·g−1,且中毒后催化剂比表面积下降,孔容也下降。以上结果表明,Ce-TiO2及Co改性后催化剂中毒后部分介孔被堵塞,硫酸铵盐的沉积或硫酸铈的生成均有可能导致这一现象。
2) TG-DTG分析。为进一步获得不同温度下SO2中毒后催化剂上硫酸铵盐、硫酸铈的定量信息,不同温度中毒后催化剂进行了TG-DTG分析,结果如图3所示。中毒后催化剂出现了3个明显的失重峰,分别始于350 ℃、490 ℃和695 ℃。纯(NH4)2SO4在280 ℃开始分解为NH4HSO4,而纯NH4HSO4在380 ℃开始分解[16]。图3中第1个失重可归结为沉积的(NH4)2SO4的分解,第2个失重峰可以归结为NH4HSO4的分解,第3个失重峰可归结为硫酸铈、硫酸钴的分解。此外,在180 ℃下Ce-TiO2催化剂沉积的硫酸铵盐失重率为1.59%,随着反应温度上升至240 ℃,硫酸铵盐失重率降低至1.1%。该结果表明,铵盐的生成随着反应温度的增加而降低。随着Co的掺杂,催化剂沉积铵盐量在从1.59%降至0.83%。该结果表明,Co的掺杂抑制了硫酸铵盐的沉积。催化剂表面沉积的铵盐在升温过程中能释放SO2,并进一步与金属氧化物反应生成硫酸盐[17]。因此,中毒后TG-DTG的高温失重峰无法为硫酸铈含量提供定量信息。
3) ICP分析。为进一步获得Ce-TiO2及Ce-Co-TiO2催化剂在不同温度下SO2中毒后催化剂上硫酸铈的定量信息,对水洗后溶液进行ICP-AES测试,结果如图4所示。在180 ℃下进行抗SO2测试,催化剂表面在最初1 h内迅速生成硫酸铈,ICP测试其Ce离子含量为0.1 mmol·g−1,经过Co掺杂后,1 h中毒后催化剂Ce离子含量下降至0.04 mmol·g−1。在240 ℃下,Ce离子含量明显增加,2 h后中毒后Ce离子含量为0.18 mmol·g−1,经过Co掺杂后,Ce离子含量下降至0.11 mmol·g−1。以上结果表明,随反应温度升高,Ce-TiO2催化剂生成的硫酸铈含量增加,Co掺杂后硫酸铈的生成被抑制。活性演变曲线表明,在180 ℃下催化剂活性迅速下降,在240 ℃下催化剂活性缓慢下降。不同温度下不同的活性演变可能揭示了不同的中毒机理及反应机理。为进一步探究180 ℃和240 ℃下Ce-TiO2催化剂不同的活性演变机理,对Ce-TiO2催化剂进行了不同时间的预硫化处理,并对预硫化后催化剂进行了ICP-AES测试和活性测试。在180 ℃ (图4 (a) 下,当硫酸铈含量达到0.1 mmol·g−1时,NO转化率对硫酸铈的沉积极其敏感,脱硝活性从50.7%迅速降至20.1%。当硫酸铈含量为0.1、0.13 mmol·g−1时,催化剂脱硝活性分别为18.5%、12.3%。因此,在180 ℃下,TG-DTG结果表明催化剂表面生成了大量的硫酸铵盐,但硫酸铵盐对Ce-TiO2催化剂低温脱硝活性的影响较小;随着1 h后硫酸铈的生成量达到0.1 mmol·g−1,低温脱硝活性快速降低;随后硫酸铈含量仍逐渐增加,低温脱硝活性则缓慢下降并最终保持在11%。随着温度升高至240 ℃,脱硝活性下降较为平缓。这可能是由于中温脱硝活性对硫酸铈的生成不是十分敏感,预硫后活性结果表明随硫酸铈含量的增加,催化剂中温活性逐渐降低,故推测在240 ℃下含硫气氛中活性的下降是由于硫酸铈的不断累积造成的。
3) XPS分析。XPS图谱可确定催化剂表面元素形态与浓度。为探究不同温度中毒对催化剂元素含量及价态的影响,对中毒前后的Ce/TiO2催化剂进行了XPS测试。图5 (a) 为不同温度中毒前后Ce 3d XPS图谱,Ce 3d XPS图谱可被拟合为8个XPS谱峰,4对Ce 3d5/2/Ce 3d3/2谱峰,从高结合能到低结合能依次标记为u’’’ (916.7 eV) 、u’’ (907.3 eV) 、u’ (903.9 eV) 、u (900.9 eV) 、v’’’ (898.3 eV) 、v’’ (888.6 eV) 、v’ (885.8 eV) 、v (882.2 eV) 。其中,u’和v’峰归属于Ce3+,其余谱峰归属于Ce4+。经过SO2中毒后,v' 和u'所对应的峰相对强度逐渐增加,而v''和 u''所对应的峰相对强度逐渐减少。随着反应温度的升高,这种现象更加明显,该现象表明,随着中毒温度升高,催化剂表面Ce3+含量逐渐升高,这可能是由于表面Ce2(SO4)3的大量生成。图5 (b) 为不同温度中毒后催化剂的S 2p图谱。167.9 eV和169.1 eV的XPS峰分别对应于S 2p3/2和S 2p1/2,这表明S6+出现在中毒后的催化剂表面。进一步对催化剂表面元素组分进行定量分析,结果见表2。随着Co的掺杂,催化剂表面Ce3+含量从25.6%增至29.1%,中毒后表面S含量则分别从3.48%、4.23%降至3.02%、3.48%。该结果表明Co掺杂后催化剂表面的S含量降低,且Ce3+/Ce比例增加量也较小,这说明Co掺杂后表面Ce2(SO4)3的生成量减少。图5 (c) 为中毒前后Co 2p的XPS谱图,根据Co 2p1/2和2p3/2轨道分裂结合能差值15.2 eV及1比2面积比例进行分峰拟合,Co3+/Co比例见表2。新鲜Ce-Co/TiO2催化剂中Co3+/Co比值为48.1%,经过抗硫中毒实验后该比例下降至46.9%和45.8%。这可能是由于部分三价Co被硫酸化生成CoSO4。
-
脱硝活性尤其是低温活性依赖于催化剂的氧化还原性能。H2-TPR测试结果如图6 (a) 所示。Ce-TiO2催化剂只显示一个还原峰,始于270 ℃且还原峰中心在455 ℃,该峰归属于CeO2的还原[18]。在Co掺杂后,2个还原峰开始出现,分别位于356 ℃和537 ℃。该结果表明Co掺杂后促进了催化剂的氧化还原性能,并因此进一步促进了催化剂的低温脱硝活性。NH3的吸附长期以来被认为是脱硝反应的第一步,NH3-TPD测试结果如图6 (b) 所示。Ce-TiO2催化剂具有2个NH3脱附峰,分别位于235 ℃和575 ℃.一般认为200 ℃以下的脱附归属于弱酸位点,300 ℃以上脱附归属于强酸位点[19]。该结果表明Ce-TiO2催化剂表面具有较强的酸性。在Co掺杂后,催化剂的NH3脱附温度和脱附强度均无明显变化,说明酸性未发生明显改变。为进一步探究SO2在Co改性前后的Ce-TiO2催化剂的吸附情况,SO2-TPD测试结果如图6 (c) 所示。Ce-TiO2催化剂具有2个SO2脱附峰,分别位于200 ℃和585 ℃。在Co掺杂后,SO2脱附温度未发生改变,但峰强度明显降低。该结果表明Co掺杂后催化剂对SO2的吸附能力明显降低。图6 (d) 为Co改性前后催化剂的O1s图谱。O1s图谱在529.3 eV、531.1 eV左右两处均具有特征峰,分别归属于晶格氧Oβ、吸附氧Oɑ。值得注意的是,吸附氧Oɑ由于其更高的活动性在氧化还原反应中表现出比Oβ更高的活性,从而促进NO氧化为NO2,有利于反应活性的提高。因此,对Oɑ/(Oɑ+Oβ)比值进行计算,结果表明Co掺杂前Oɑ/(Oɑ+Oβ)为0.31,Co掺杂后该比值增加至0.43。文献[20]表明,吸附氧Oɑ可被归结于氧缺陷位或表面羟基基团,结合上述Co掺杂后Ce3+含量的增加,推断Co的掺杂导致了Ce4+向Ce3+的还原,在这一过程中晶格氧的去除导致了缺陷和氧空位的形成。
-
为进一步探究低温下硫酸铈的生成对催化剂反应机理的影响,利用原位红外技术 (in situ DRIFT) 探究催化剂反应机理的变化。首先Ce-TiO2和Ce-Co-TiO2催化剂经过180 ℃下的预硫化处理,然后放置于原位红外反应池中,预吸附NH3直至饱和后通入NO+O2,红外图谱如图7 (a) 和 (b) 所示。其中,1 184 cm−1处的振动峰可归结于吸附在L酸位点上的NH3,1 689 cm−1处的振动峰可归结于吸附在B酸位点上的NH3,1 600 cm−1可归结于NH3的过度氧化导致的硝酸盐中间产物的振动峰[21]。图7 (a) 表明,当通入NO+O2 10 min后,在1 305 cm−1处出现了新的吸附峰,这可以归因于吸附后形成的NO3−[22]。随NO+O2通入时间的增加,1 184 cm−1处的振动峰强度未发生变化。该结果表明预硫化后的Ce-TiO2表面吸附的NH3不具反应活性,脱硝反应E-R路径无法进行。图7 (b) 表明,对于预硫后的Ce-Co-TiO2催化剂,随NO+O2通入时间的增加,1 184 cm−1处的振动峰强度明显降低。该结果表明预硫后的Ce-Co-TiO2催化剂表面吸附的NH3具有反应活性,遵循E-R脱硝反应路径。
为进一步探究2种预硫后催化剂的L-H脱硝反应路径,预吸附NO+O2直至饱和随后通入NH3的红外实验被进行。图7 (c) 表明预硫后Ce-TiO2催化剂吸附NO+O2出现了1 238 cm−1、1 566 cm−1、1 600 cm−1的吸附峰,分别归属于单齿硝酸盐 (O-N-O) [23]、双齿硝酸盐 (-O-N-O-) [24]、吸附态NO2。随着NH3的通入时间增加,表面硝酸盐振动峰强度无明显变化,NH3的吸附峰开始出现。该结果表明吸附态的NH3与亚硝酸盐/硝酸盐在预硫后催化剂表面可以共存,预硫后Ce-TiO2催化剂不遵循L-H脱硝反应路径。如图7 (d) 所示,对于预硫后的Ce-Co-TiO2催化剂,1 331 cm−1和1 620 cm−1处的振动峰可分别归属于单齿硝酸盐 (O-N-O) 和NO2,当通入20 min的NH3进行反应,1 331 cm−1和1 620 cm−1处的振动峰完全消失。这表明预硫后Ce-Co-TiO2催化剂表面上吸附态的硝酸盐能与NH3反应生成NH4NO3,并进一步分解生成N2和H2O[25],预硫后Ce-Co-TiO2催化剂能够遵循L-H路径进行脱硝反应。
-
1) 在180 ℃下,由于低温脱硝活性对催化剂氧化还原性能具有较高的要求,较低含量硫酸铈的生成就会导致低温脱硝活性的急剧下降。随着反应温度升高至240 ℃,催化剂表面硫酸铈生成量明显增加,催化活性缓慢下降,中温脱硝活性对硫酸铈的生成敏感性较低,持续的硫酸铈生成导致了催化活性的持续缓慢下降。
2) Co改性能在一定程度上提升Ce-TiO2催化剂的脱硝活性及中低温抗硫活性。这主要是由于Co掺杂提升了催化剂的氧化还原性能,且抑制了SO2在催化剂表面的吸附,进而维持了催化剂E-R、L-H脱硝反应路径的进行。
Ce-TiO2催化剂在含SO2气氛下的NH3-SCR中毒机理及其Co3O4改性性能
Study on NH3-SCR poisoning mechanism and Co3O4 modification of Ce-TiO2 catalyst in SO2 atmosphere
-
摘要: 通过对Ce-TiO2催化剂在含硫气氛下、不同温度的NH3-SCR活性演变,结合含硫组分的定量分析、原位红外分析,研究了Ce-TiO2催化剂在不同温度的SO2中毒机理。结果表明,在180 ℃下,Ce-TiO2脱硝活性对硫酸铈的生成极为敏感,0.1 mmol·g−1的硫酸铈生成导致脱硝活性从50.7%降至18.5%,随后硫酸铈持续沉积,低温脱硝活性缓慢下降;在240 ℃下,Ce-TiO2脱硝活性对硫酸铈的生成敏感性较低,脱硝活性随时间缓慢下降,0.18 mmol·g−1的硫酸铈生成导致脱硝活性从100%降至53.8%,随后硫酸铈持续生成。Co改性活性结果表明:在180 ℃时,脱硝活性从50.7%提升至94.2%,且180 ℃和240 ℃抗硫性能均有所提升。进一步的表征测试表明:Co的掺杂能提升Ce-TiO2催化剂的氧化还原性能,并抑制SO2在催化剂表面的吸附,提升催化剂的抗硫性能。原位红外测试结果表明:硫酸化后的Ce-Co-TiO2催化剂仍能维持了E-R、L-H脱硝反应路径的进行,保证了一定的低温脱硝活性。本研究探索了SCR催化剂的中毒机制,可为其改性与活性提升提供参考。
-
关键词:
- 氨气选择性催化还原(NH3-SCR) /
- 脱硝 /
- 催化剂 /
- 硫酸铈
Abstract: The SO2 poisoning mechanism of the Ce-TiO2 catalyst at different temperatures was investigated by the evolution of NH3-SCR activity under SO2-containing atmosphere, combined with the quantitative analysis of sulfur-containing components and in-situ DRIFTS analysis. The results demonstrated that the denitrification activity of Ce-TiO2 was extremely sensitive to the generation of cerium sulfate at 180 ℃, and the generation of 0.1 mmol·g−1 of cerium sulfate led to the rapid decrease of denitrification activity from 50.7% to 18.5%, followed by the continuous deposition of cerium sulfate and the slow decrease of low-temperature denitrification activity. When the reaction temperature increased to 240 ℃, the denitrification activity of Ce-TiO2 was less sensitive to the generation of cerium sulfate, and the denitrification activity slowly decreased from 100% to 53.8% with the generation of 0.18 mmol·g−1 cerium sulfate. With the doping of Co3O4, the denitrification activity was increased from 50.7% to 94.2% at 180 °C, and the sulfur resistance was improved at both 180 °C and 240 °C. Further characterization tests showed that Co doping could enhance the redox performance of the Ce-TiO2 catalyst and inhibited the adsorption of SO2 on the catalyst surface to enhance the sulfur resistance of the catalyst. The in-situ infrared spectra demonstrated that the sulfated Ce-Co-TiO2 catalyst could still maintain the E-R and L-H denitrification reaction paths and ensure a certain low-temperature denitrification activity. -
-
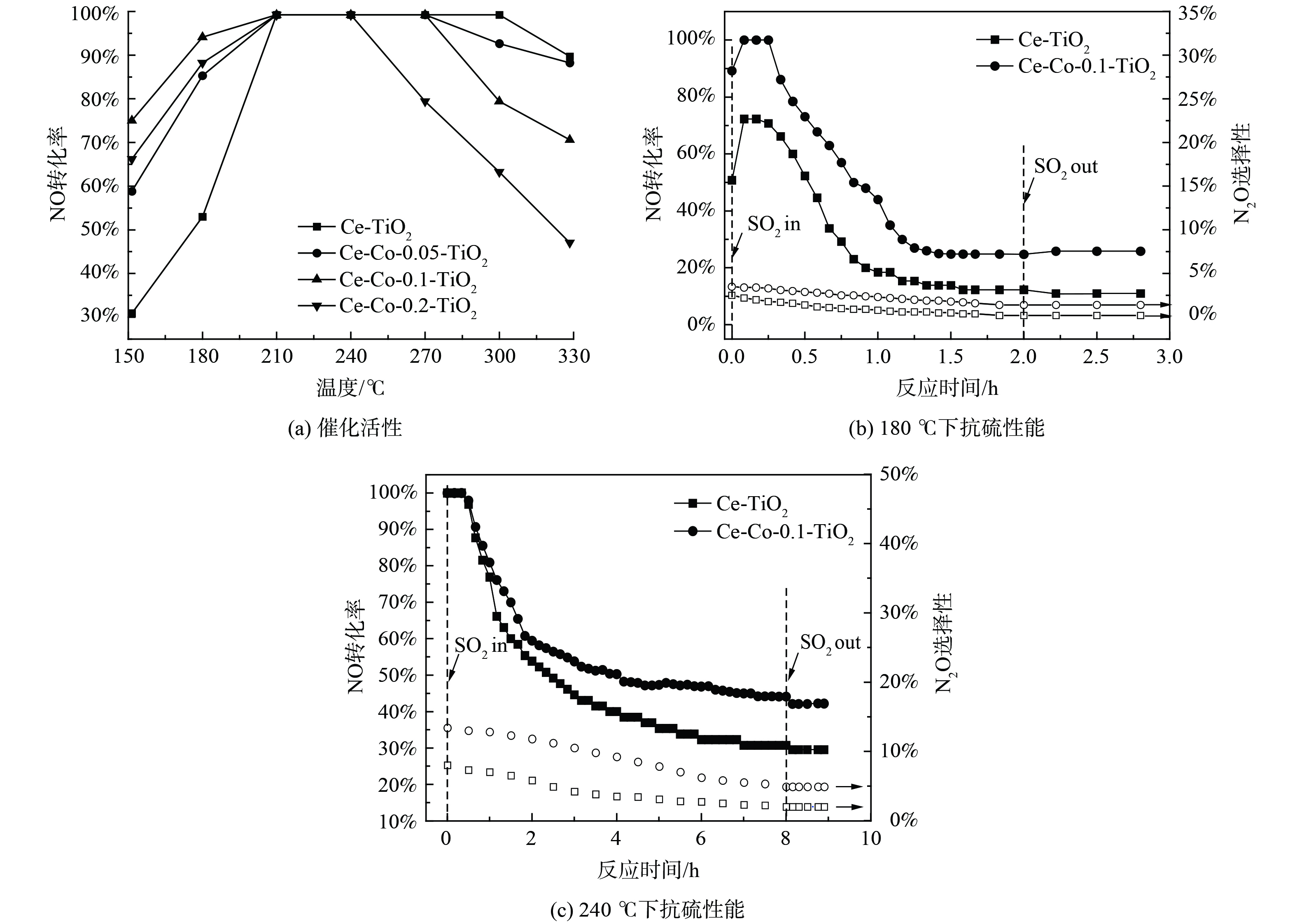
图 1 Ce-(Co)-TiO2催化剂的NO转化率及抗硫性能
Figure 1. NO conversion and sulfur resistance of Ce-(Co)-TiO2 catalyst
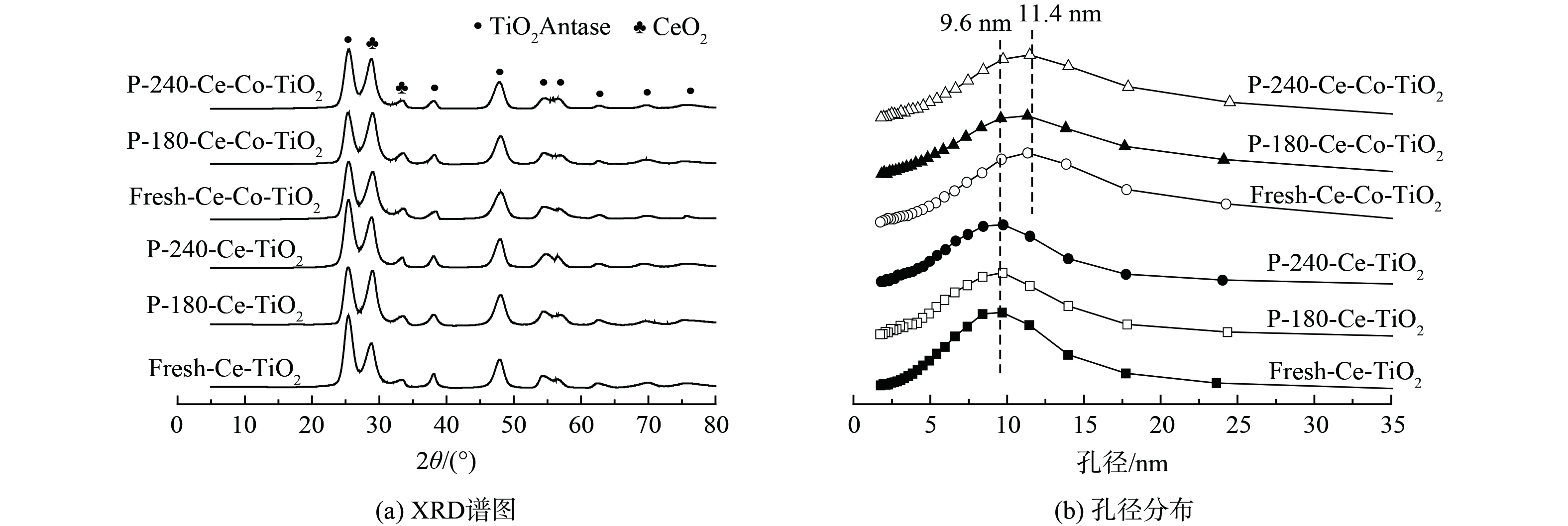
图 2 新鲜以及不同温度中毒后Ce-(Co)-TiO2催化剂的XRD谱图与孔径分布
Figure 2. XRD patterns and pore size distribution of fresh Ce-(Co)-TiO2 catalysts and Ce-(Co)-TiO2 catalysts poisoned under different temperatures
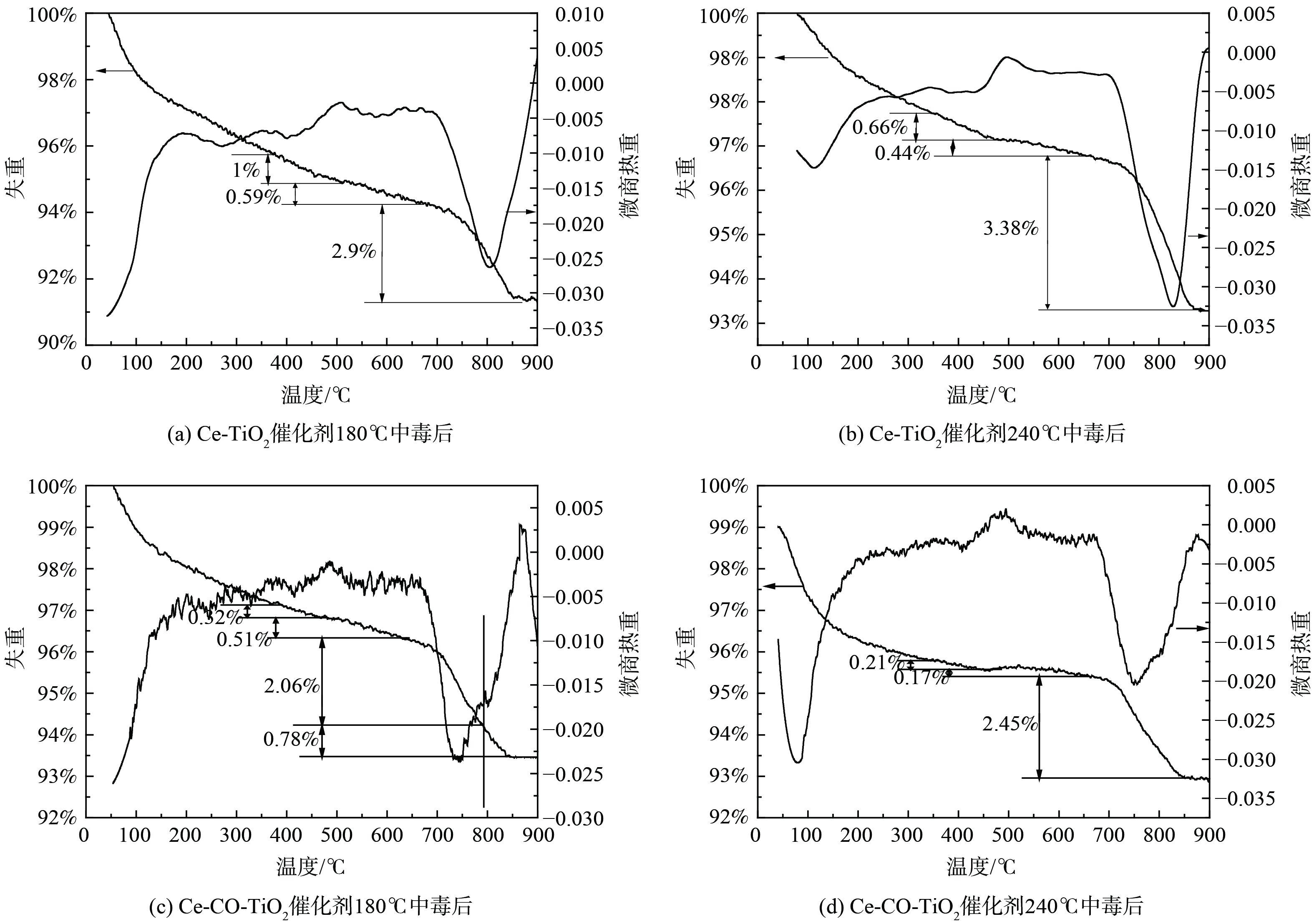
图 3 Ce-(Co)-TiO2催化剂2h中毒后TG-DTG曲线
Figure 3. TG-DTG profiles of the SO2-poisoned Ce-(Co)-TiO2 catalysts for 1h.
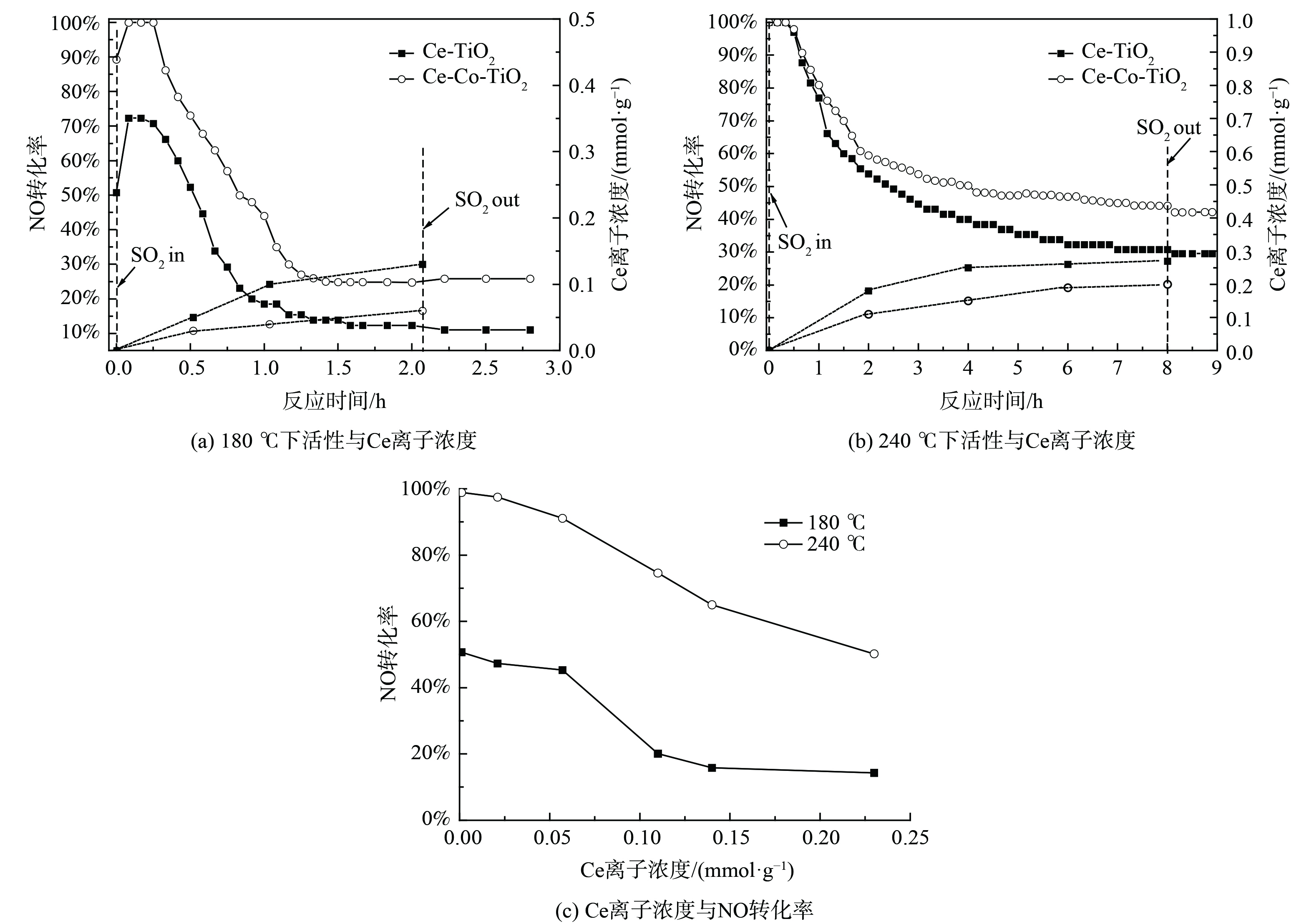
图 4 SO2存在条件下催化剂活性与铈离子含量关系
Figure 4. Relationship between catalytic activity and Ce ion concentration in the presence of SO2
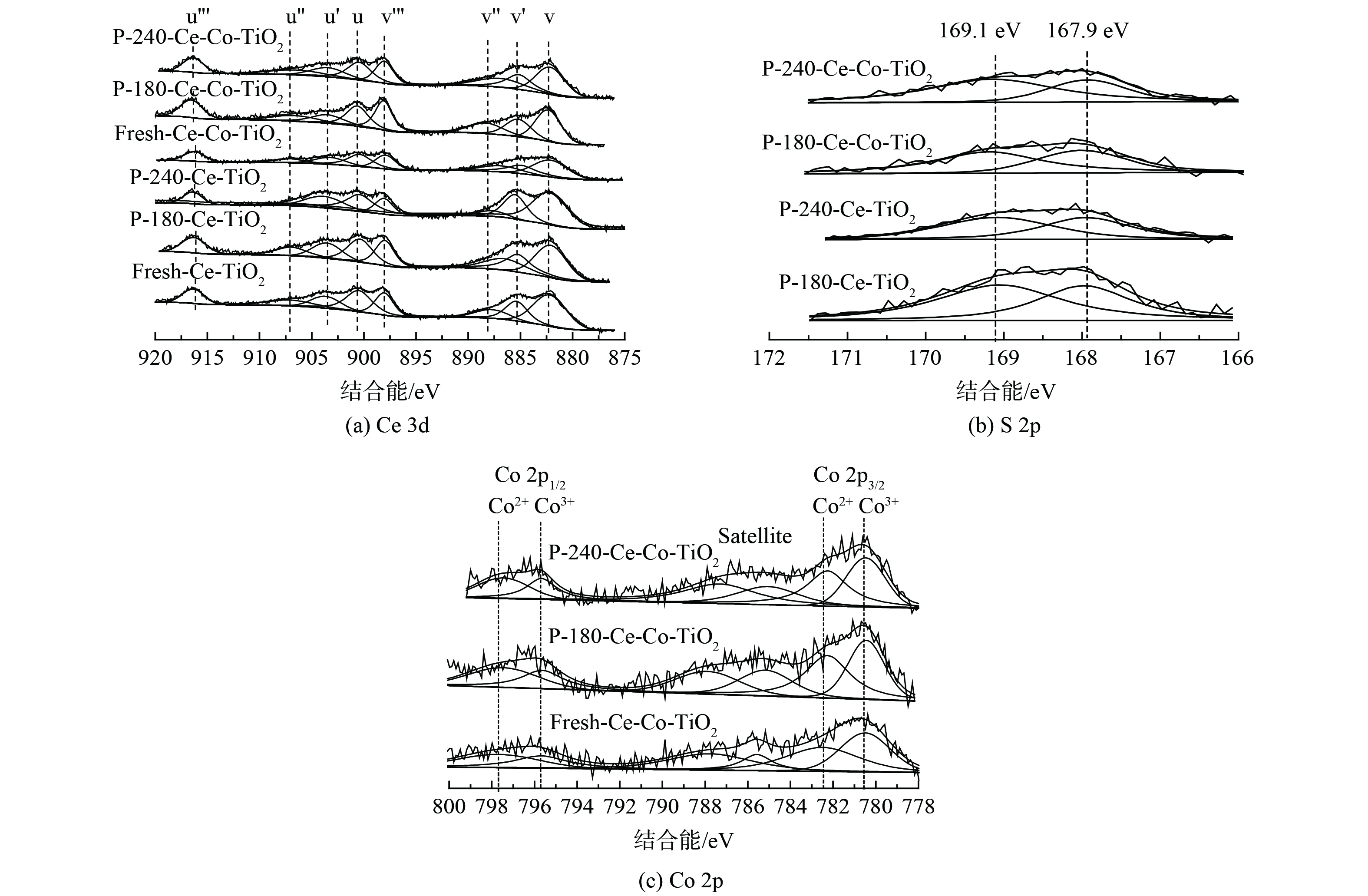
图 5 不同温度中毒前后XPS图谱
Figure 5. XPS spectra of Ce-TiO2 and Ce-Co-TiO2 before and after SO2 poisoning

图 6 Co掺杂前后Ce-TiO2催化剂物化特性表征
Figure 6. Physiochemical properties of Ce-TiO2 catalysts with/without Co doping

图 7 中毒前后Ce-(Co)-TiO2催化剂在180 ℃下预吸附气体后反应的原位红外图谱
Figure 7. In-situ infrared spectra of Ce-(Co)-TiO2 catalysts before and after preadsorption of gas at 180 ℃
表 1 新鲜以及中毒后Ce-TiO2及Ce-Co-TiO2催化剂的结构参数
Table 1. Structural parameters of fresh and poisoned Ce-TiO2 and Ce-Co-TiO2 catalysts
样品名称 Sbet/(m2·g−1) Vt/(cm3·g−1) Fresh Ce-TiO2 120.9 0.24 P-180-Ce-TiO2 109.4 0.20 P-240-Ce-TiO2 95.1 0.18 Fresh Ce-Co-TiO2 98.7 0.22 P-180-Ce-Co-TiO2 91.6 0.20 P-240-Ce-Co-TiO2 90.1 0.20  下载: 导出CSV
下载: 导出CSV
表 2 新鲜及中毒后Ce-(Co)-TiO2表面原子含量及价态比例
Table 2. Atomic content and valence state ratio of Ce-(Co)-TiO2 on fresh and poisoned surface
样品名称 S元素含量 比例 Ce3+/Ce Co3+/Co Fresh Ce-TiO2 — 25.6% — P-180-Ce-TiO2 3.48% 34.5% — P-240-Ce-TiO2 4.23% 41.9% — Fresh Ce-Co-TiO2 — 29.1% 48.1% P-180-Ce-Co-TiO2 3.02% 32.4% 46.9% P-240-Ce-Co-TiO2 3.48% 34.7% 45.8%
下载: 导出CSV
-
[1] FORZATTI P. Environmental catalysis for stationary applications[J]. Catalysis Today, 2000, 62(1): 51-65. doi: 10.1016/S0920-5861(00)00408-9 [2] MACHIDA M, MURATA Y, KISHIKAWA K, et al. On the reasons for high activity of CeO2 catalyst for soot oxidation[J]. Chemistry of Materials, 2008, 20(13): 4489-4494. doi: 10.1021/cm800832w [3] XIAO X, XIONG S, SHI Y, et al. Effect of H2O and SO2 on the selective catalytic reduction of NO with NH3 over Ce/TiO2 catalyst: mechanism and kinetic study[J]. The Journal of Physical Chemistry C, 2016, 120(2): 1066-1076. doi: 10.1021/acs.jpcc.5b10577 [4] SHEN Y, ZHU S, QIU T, et al. A novel catalyst of CeO2/Al2O3 for selective catalytic reduction of NO by NH3[J]. Catalysis Communications, 2009, 11(1): 20-23. doi: 10.1016/j.catcom.2009.08.001 [5] JIN Q, SHEN Y, ZHU S, et al. Rare earth ions (La, Nd, Sm, Gd, and Tm) regulate the catalytic performance of CeO2/Al2O3 for NH3-SCR of NO[J]. Journal of Materials Research, 2017, 32(12): 2438-2445. doi: 10.1557/jmr.2017.125 [6] FRANCE L J, YANG Q, LI W, et al. Ceria modified FeMnOx-Enhanced performance and sulphur resistance for low-temperature SCR of NOx[J]. Applied Catalysis B:Environmental, 2017, 206: 203-215. doi: 10.1016/j.apcatb.2017.01.019 [7] LIU Z, ZHU J, LI J, et al. Novel Mn-Ce-Ti mixed-oxide catalyst for the selective catalytic reduction of NOx with NH3[J]. ACS Applied Materials & Interfaces, 2014, 6(16): 14500-14508. [8] ZHANG L, LI L, CAO Y, et al. Getting insight into the influence of SO2 on TiO2/CeO2 for the selective catalytic reduction of NO by NH3[J]. Applied Catalysis B:Environmental, 2015, 165: 589-598. doi: 10.1016/j.apcatb.2014.10.029 [9] XU W, HE H, YU Y. Deactivation of a Ce/TiO2 catalyst by SO2 in the selective catalytic reduction of NO by NH3[J]. The Journal of Physical Chemistry C, 2009, 113(11): 4426-4432. doi: 10.1021/jp8088148 [10] KWON D W, NAM K B, HONG S C. The role of ceria on the activity and SO2 resistance of catalysts for the selective catalytic reduction of NOx by NH3[J]. Applied Catalysis B:Environmental, 2015, 166: 37-44. [11] XU L, WANG C, CHANG H, et al. New insight into SO2 poisoning and regeneration of CeO2-WO3/TiO2 and V2O5-WO3/TiO2 catalysts for low-temperature NH3-SCR[J]. Environmental Science & Technology, 2018, 52(12): 7064-7071. [12] HUANG J, TONG Z, HUANG Y, et al. Selective catalytic reduction of NO with NH3 at low temperatures over iron and manganese oxides supported on mesoporous silica[J]. Applied Catalysis B:Environmental, 2008, 78(3-4): 309-314. doi: 10.1016/j.apcatb.2007.09.031 [13] MA Z, YANG H, LI B, et al. Temperature-dependent effects of SO2 on selective catalytic reduction of NO over Fe-Cu-Ox/CNTs-TiO2 Catalysts[J]. Industrial & Engineering Chemistry Research, 2013, 52(10): 3708-3713. [14] JIN R B, LIU Y, WU Z, et al. Relationship between SO2 poisoning effects and reaction temperature for selective catalytic reduction of NO over Mn-Ce/TiO2 catalyst[J]. Catalysis Today, 2010, 153(3/4): 84-89. [15] LIU F, ASAKURA K, HE H, et al. Influence of sulfation on iron titanate catalyst for the selective catalytic reduction of NOx with NH3[J]. Applied Catalysis B:Environmental, 2011, 103(3/4): 369-377. doi: 10.1016/j.apcatb.2011.01.044 [16] ZHU Z, NIU H, LIU Z, et al. Decomposition and reactivity of NH4HSO4 on V2O5/AC catalysts used for NO reduction with ammonia[J]. Journal of Catalysis, 2000, 195(2): 268-278. doi: 10.1006/jcat.2000.2961 [17] MA Z, WU X, FENG Y, et al. Low-temperature SCR activity and SO2 deactivation mechanism of Ce-modified V2O5-WO3/TiO2 catalyst[J]. Progress In Natural Science:Materials International, 2015, 25(4): 342-352. doi: 10.1016/j.pnsc.2015.07.002 [18] MURUGAN B, RAMASWAMY A V. Chemical states and redox properties of Mn/CeO2-TiO2 nanocomposites prepared by solution combustion route[J]. The Journal of Physical Chemistry C, 2008, 112(51): 20429-20442. doi: 10.1021/jp806316x [19] LIU Z, ZHANG S, LI J, et al. Novel V2O5-CeO2/TiO2 catalyst with low vanadium loading for the selective catalytic reduction of NOx by NH3[J]. Applied Catalysis B:Environmental, 2014, 158: 11-19. [20] KANG M, PARK E D, KIM J M, et al. Manganese oxide catalysts for NOx reduction with NH3 at low temperatures[J]. Applied Catalysis A:General, 2007, 327(2): 261-269. doi: 10.1016/j.apcata.2007.05.024 [21] LARRUBIA M A, RAMIS G, BUSCA G. An FT-IR study of the adsorption of urea and ammonia over V2O5-MoO3-TiO2 SCR catalysts[J]. Applied Catalysis B:Environmental, 2000, 27(3): L145-L151. doi: 10.1016/S0926-3373(00)00150-8 [22] RUGGERI M P, SELLERI T, COLOMBO M, et al. Investigation of NO2 and NO interaction with an Fe-ZSM-5 catalyst by transient response methods and chemical trapping techniques[J]. Journal of Catalysis, 2015, 328: 258-269. doi: 10.1016/j.jcat.2015.02.003 [23] PENG Y, LI J, HUANG X, et al. Deactivation mechanism of potassium on the V2O5/CeO2 catalysts for SCR reaction: acidity, reducibility and adsorbed NOx[J]. Environmental Science & Technology, 2014, 48(8): 4515-4520. [24] MARTINEZ A A, SORIA J, CONESA J C, et al. NO reaction at surface oxygen vacancies generated in cerium oxide[J]. Journal of The Chemical Society, Faraday Transactions, 1995, 91(11): 1679-1687. doi: 10.1039/FT9959101679 [25] NOVA I, CIARDELLI C, TRONCONI E, et al. NH3-NO/NO2 chemistry over V-based catalysts and its role in the mechanism of the fast SCR reaction[J]. Catalysis Today, 2006, 114(1): 3-12. doi: 10.1016/j.cattod.2006.02.012 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2929
- HTML全文浏览数: 2929
- PDF下载数: 124
- 施引文献: 0
