

 下载:
下载:

-
铬污染主要来源于采矿、金属加工、电镀、印染等工业废水,主要以Cr(VI)的形式长期稳定存在于生态系统中[1-2]。亚硫酸盐(S(IV))是常用的工业还原剂之一,能有效还原Cr(VI)(式(1)~(3))[3],并且亚硫酸盐还原Cr(VI)技术已在实际工程广泛使用(《钢铁行业采选矿工艺污染防治最佳可行技术指南(试行)》HJ-BAT-003)。有研究[4-5]表明,S(IV)与Cr(VI)的反应过程中会产生一系列活性自由基,如SO3·−、SO4·−和SO5·−等(式(4)~(10)),可用于氧化废水中共存的抗生素、酚类和染料等有机污染物,对废水进行同步氧化和还原处理[6-8]。
然而,在S(IV)活化过程中,SO32−/HSO3-极易与产生的自由基SOx·−发生副反应,造成自由基内耗(式(8)~(10)),从而抑制S(IV)体系中污染物的氧化。前期的研究[13]表明,葡萄糖基碳材料可以催化活化S(IV)产生硫氧自由基氧化污染物,其活化可能是通过S(IV)与碳材料上的官能团络合并发生分子内电子移动完成的。如果将葡萄糖基碳材料引入Cr(VI)/S(IV)体系,Cr(VI)与S(IV)反应生成CrSO62−,并有可能与碳材料表面官能团发生配位,生成的不稳定络合物自分解产生自由基[14-16],从而强化Cr(VI)/S(IV)体系对污染物的氧化降解作用,同时在空间上抑制与SOx·−的副反应,减少自由基内耗,为强化Cr(VI)/S(IV)体系降解有机污染物提供思路。
基于此,本研究采用双氯芬酸(diclofenac,DCF)为目标污染物,研究葡萄糖衍生碳材料强化Cr(VI)/S(IV)体系氧化降解污染物的性能,并深入探究了其促进机理。以期为Cr(VI)和S(IV)的有效利用,以及碳材料强化Cr(VI)/S(IV)体系的实际应用提供参考。
-
1)实验试剂。葡萄糖(AR),亚硫酸盐(Na2SO3,98%),重铬酸钾(K2Cr2O7),双氯芬酸钠(DCF,99%),甲醇((methanol,MeOH,99.5%,AR),NaOH(99%,AR),H2SO4(95%~98%),乙酸(99.5%,AR),二苯碳酰二肼(99%,AR),丙酮(99.5%,AR),乙二胺四乙酸(Ethylenediaminetetraacetic acid,EDTA,99%,AR),磷酸氢二钾(99%,AR),磷酸二氢钾(99%,AR)和叔丁醇(tert-butyl alcohol,TBA,99.5%,AR)等购自国药集团化学试剂有限公司(中国上海),乙腈(HPLC级)购自赛默飞世尔科技(中国)有限公司,过硫酸铵(AP,99.99%,AR)和5,5-二甲基-吡啶N-氧化物(5,5-dimethyl-1-pyrroline N-oxide,DMPO,97%)购自上海阿拉丁,5,5-二硫代双-(2-硝基苯甲酸)( 5,5’-Dithiobis-(2-nitrobenzoic acid),DTNB,98%,AR)购自Sigma-Aldrich。本研究中的所有化学药品和试剂均按原样使用,无需进一步纯化。
2)实验仪器。高温管式炉(SX-2-5-10,武汉电炉厂),电子分析天平(BSA124S-CW,德国赛多利斯(Sartorius)),恒温鼓风干燥箱(DZG-6 020,天津华北实验有限公司),纯水仪(PCDX-J2-10,成都品成科技有限公司),恒温磁力搅拌水浴锅(ZNCL-S,科尔仪器设备有限公司),UV-Vis分光光度计(UV-2 600,日本岛津(Shimadzu)),高效液相色谱(HPLC LC-15C,日本岛津(Shimadzu))。
-
以葡萄糖为前驱体,在氮气气氛下以3 ℃·min−1的升温速率,于1 000 ℃煅烧2 h,冷却至室温,将得到的碳材料记为C-1000。
氧化处理:使用湿式化学氧化法[17-18]对碳材料进行氧化处理。处理过程如下:将过量的过硫酸铵添加到1 mol·L−1的H2SO4溶液中制备饱和过硫酸铵溶液。称取0.5 g碳材料搅拌分散到50 mL氧化溶液中,经过硫酸铵氧化一定时间后用大量去离子水冲洗过滤,并于60 ℃下干燥24 h。获得的氧化材料记为OC-t(t表示氧化时间,h)。
还原处理:使用水热还原法[19]对碳材料进行还原处理。处理过程如下:称取0.2 g碳材料加入40 mL水中,通过1 mol·L−1 NaOH溶液将pH调节至11,超声30 min并搅拌30 min后,将得到的混合物转移至内衬特氟隆的高压釜(50 mL)中,分别在160 ℃和200 ℃加热2 h,自然冷却至室温后取出,使用大量去离子水冲洗过滤,并于60 ℃下干燥24 h。获得的还原材料RC-T(T表示水热还原的温度, ℃)。
-
实验在150 mL烧杯中进行。实验过程如下:取适量Na2SO3储备液和DCF储备液配制50 mL反应液,使用稀H2SO4将pH调节至7。加入适量K2Cr2O7储备液和催化剂引发反应,其中Na2SO3浓度为2.4 mmol·L−1,DCF的浓度为15 μmol·L−1,Cr(VI)浓度为0.04 mmol·L−1,C-1000投加量为0.1 g·L−1 (若无特殊标注,则反应条件与上述一致) 。在给定的时间间隔内,取2 mL悬浮液并用0.22 μm水系滤头过滤以进行进一步分析。DCF浓度通过高效液相色谱进行分析,流动相为乙腈/水(50:50),流速0.8 mL·min−1,在276 nm处测定。为防止取样后反应继续进行影响DCF浓度测定,在0.9 mL滤液中加入0.1 mL甲醇以终止反应。溶液中Cr(VI)的测定采用二苯碳酰二肼显色法,于540 nm波长处测定吸光度。S(IV)的含量使用DTNB比色法测定,在412 nm波长处测定吸光度。反应过程中产生的活性自由基使用DMPO作为自由基的自旋捕集剂,通过EPR光谱仪进行检测。实验在恒温水浴条件下进行,控制温度在(25±1) ℃,所有实验均重复3次,结果的平均值相对误差小于±5%。
不同体系的对比实验:向DCF溶液、S(IV)/DCF混合溶液、Cr(VI)/DCF混合溶液中加入C-1000催化剂,在相同条件下进行C-1000/DCF、C-1000/S(IV)/DCF、C-1000/Cr(VI)/DCF体系的对照实验。向S(IV)/DCF混合溶液中,投加K2Cr2O7储备液,进行Cr(VI)/S(IV)/DCF体系对照实验。
不同气氛实验:向一定量纯水中进行30 min的预曝气(N2、Air、O2),之后依次加入适量Na2SO3储备液、DCF储备液、稀硫酸、K2Cr2O7储备液及催化剂引发反应,保持曝气,并按照取样时间进行单瓶实验。
淬灭实验:在反应开始前,向S(IV)-DCF混合溶液中分别加入0、2、5、10、20 mmol·L−1的甲醇作为自由基淬灭剂,混合均匀后加入适量K2Cr2O7储备液和催化剂引发反应,进行甲醇淬灭实验;按照相同的操作步骤,以叔丁醇作为·OH淬灭剂进行淬灭实验。
-
用场发射扫描电子显微镜(FEI-Sirion 200,荷兰FEI公司)研究催化剂的微观形貌。使用EPR(EMXnano,德国布鲁克公司)光谱仪检测反应过程中形成的自由基。通过X射线衍射仪(x’pert3 powder,荷兰PANalytical B.V.公司)检查获得的样品的相和晶体结构。使用傅立叶变换红外(Nicolet iS50R,美国赛默飞世尔科技公司)光谱分析材料的表面官能团。使用X射线光电子能谱法(AXIS-ULTRA DLD-600 W,日本岛津(Shimadzu))对材料表面反应前后的官能团变化进行分析。通过拉曼光谱(LabRAM HR800,法国HORIBA Jobin Yvon公司)评估催化剂的缺陷程度。
-
如图1(a)所示,C-1000材料表现为典型的碳材料无定型块状结构,表面无孔且相对光滑。由图1(b)中 XRD的表征结果可见,在23°和43.5°处出现2θ的2个宽峰[20],分别代表结晶碳的(002)和(100)平面。由图1(c)中FT-IR图谱可见,在1 013、1 155、1 631、1 713、2 911 cm−1处的特征峰分别归属于C—O、C—O—C、C=O、O—C=O和C—H的拉伸振动峰,在3 438 cm−1处观察到的宽峰可归属于—OH或水分子的拉伸振动峰[21-22]。根据拉曼光谱(图1(d)),C-1000的ID/IG值为1.23,D波段和G波段分别在1 357 cm−1和1 610 cm−1处[23]。
-
图2(a)反映了不同体系对DCF的降解效果。结果表明,C-1000/Cr(VI)/S(IV)体系的DCF降解率可达到100%,而在不加C-1000的情况下,Cr(VI)/S(IV)体系中的DCF去除率仅为65%。为了探究C-1000的吸附作用,单独加入C-1000进行对比实验,结果表明,C-1000对DCF几乎不产生吸附。以C-1000/S(IV)体系和C-1000/Cr(VI)体系作为对照,探究C-1000的单独活化作用,结果表明,DCF的去除率不到5%。因此,C-1000能够有效增强Cr(VI)/S(IV)体系对有机物的氧化降解效能。
通过Cr(VI)还原曲线以及S(IV)分解曲线,探究C-1000对Cr(VI)/S(IV)体系降解效果的影响。如图2(b)所示,反应开始10 min时,Cr(VI)/S(IV)/DCF体系仅有10%的Cr(VI)被还原。这归因于在反应初期,S(IV)有一定的pH缓冲能力,Cr(VI)在偏中性条件下主要以HCrO4−形式存在,氧化能力较弱[24];C-1000加入后显著加快了Cr(VI)的还原速率。结合图2(a)、图2(b)可见,DCF的降解速率变化与Cr(VI)还原率变化趋势相吻合。S(IV)的分解曲线如图2(c)所示,加入C-1000后S(IV)的分解速率加快,最终消耗量却有所减少,这也表明在C-1000强化过程中S(IV)的有效利用率得到进一步提高,侧面验证了活性自由基增加的可能性。
-
1) S(IV)浓度的影响。S(IV)由于自身的氧化还原特性,一方面被活化产生自由基,另一方面可作为还原剂淬灭自由基,从而导致降解效果变差,因此,研究S(IV)的投加量至关重要。如图3(a)所示,当S(IV)浓度由0.8 mmol·L−1增加至2.4 mmol·L−1时,DCF去除率明显提高,反应60 min时DCF去除率由30%提高到100%。这主要是因为当S(IV)浓度升高后,体系中产生的活性自由基也随之增加,从而使得DCF降解效率得到大幅提升。但当S(IV)浓度进一步提高至3.2 mmol·L−1时,过量的S(IV)与自由基反应,与DCF造成竞争效应,导致降解效果变差。
2) Cr(VI)浓度的影响。Cr(VI)作为强氧化剂,在整个亚硫酸盐体系中起到激活氧化的作用。本研究考察了Cr(VI)投加量对C-1000/Cr(VI)/S(IV)体系降解DCF的影响。如图3(b)所示,DCF的降解率随着Cr(VI)浓度增加而增加,Cr(VI)浓度由0.01 mmol·L−1提高为0.04 mmol·L−1时,DCF的降解速率和去除率均有大幅提高。这归因于S(IV)被进一步活化,自由基的产生速率加快。但当Cr(VI)浓度继续增加,DCF的去除速率的提升不明显,这表明此时自由基的产生速率已经达到最大,S(IV)浓度成为提高反应速率的限制因素。
3) C-1000投加量的影响。进一步研究了催化剂投加量对DCF降解效率的影响。如图3(c)所示,增加C-1000的投加量能显著提高DCF的降解效率。催化剂投加量为0.05 g·L−1时,DCF的去除率为86%;当催化剂投加量增加至0.2 g·L−1时,DCF的去除率增加至100%,同时反应速率常数从0.083 6 min−1提高至0.273 6 min−1。这可能是因为C-1000投加量的增加可为体系提供更多活性位点,有利于产生更多的活性自由基,进而提高对DCF的降解效率。综合考虑高效性和经济性,选择0.1 g·L−1作为本体系中催化剂投加量。
-
上述结果表明,C-1000可以显著增强Cr(VI)/S(IV)体系的氧化性能。有研究表明,在S(IV)活化体系,活性物种主要包括SO3·−、SO4·−、SO5·−和·OH[3,8],因此,推测C-1000有利于活性自由基放入产生。据报道,甲醇(MeOH)可以同时淬灭SO3·−(k = 1.6×104 L·(mol·s)−1)、SO4·−(k = 3.2×106 L·(mol·s)−1)和·OH(k = 9.7×108 L·(mol·s)−1)[25],而叔丁醇(TBA)通常用于淬灭·OH(k = 3.8×108 ~7.6×108 L·(mol·s)−1)[26]。因此,选用MeOH和TBA作为自由基淬灭剂分析反应过程中的自由基种类和贡献。如图4(a)和图4(b)所示,在C-1000/Cr(VI)/S(IV)体系中,过量MeOH的加入几乎完全抑制了DCF的降解,而加入过量TBA仅有部分抑制。该结果证明在C-1000/Cr(VI)/S(IV)体系中,SO5·−的作用可忽略不计,因为SO5·−对醇是惰性的(k< 103 L·(mol·s)−1)[8];·OH对DCF的氧化有部分贡献,但并不是起主要作用的活性物种。
设计气氛实验进一步确定SO3·−和SO4·−的作用。在由SO3·−引发的链式自由基反应中,产生SO4·−的关键在于O2的参与,其中SO5·−是SO4·−的前体。如图4(c)所示,在N2饱和体系中,反应30 min时DCF的氧化率仅为5%;而在O2饱和体系中,30 min时DCF去除率已达到98%。该现象说明溶液中的O2是DCF氧化降解的重要影响因素。在氮气饱和体系中,由于溶液中溶解氧含量极低,SO3·−无法进一步反应生成SO4·−和SO5·−,导致DCF降解几乎被完全抑制;而在氧饱和系统中,O2可与SO3·−反应生成SO4·−,从而加快DCF的氧化降解。上述实验结果表明,在C-1000/Cr(VI)/S(IV)体系氧化降解DCF过程中,SO4·−起主要作用。
如图4(d)所示,在相同反应时间下,对比Cr(VI)/S(IV)体系,C-1000/Cr(VI)/S(IV)体系中DMPO/SO3·−加合物的信号峰强度明显增加,这表明C-1000能够显著促进Cr(VI)/S(IV)体系中SO3·−的产生。图4(e)反映了C-1000/Cr(VI)/S(IV)体系在反应开始前10 min内SO3·−的产生数量情况。如图4(e)所示,DMPO/SO3·−峰强度随反应时间呈现先升高后降低的趋势,在反应3 min时峰强度最高,此时C-1000催化活化Cr(VI)/S(IV)体系产生的SO3·−达到最高,SO3·−与O2反应引发链式反应,产生了更多活性更强的SO4·−,从而促进DCF氧化。在图4(d)、图4(e)中未检测到SO4·−的信号,可以归因于在S(IV)活化体系中,SO4·−主要由SO5·−与HSO3−或SO5·−反应生成,根据反应速率常数,SO5·−与SO5·−的反应速率明显高于HSO3−(式(7)~(8)),但由于HSO3−与SO4·−存在副反应,其反应速率常数与式(8)相当(均为108),从而降低了SO4·−的有效利用率。另一方面,很可能是因为过量的DMPO可捕获所有的SO3·−并终止随后的自由基链反应[6],并且DMPO-SO4·−加合物的灵敏度较低,一般来说较难检测到,在其他S(IV)活化体系中也有类似的结果[25],因而没有观察到明显的DMPO-SO4·−加合物信号。而溶液中已存的SO4·−易转化为SO42−与HSO4−等更加稳定的离子(式(5)~(6))。
-
有研究表明,多羧酸盐的羧基(—COOH)可以与CrSO62−络合从而形成不稳定的络合物,通过电子内部转移可以快速自分解产生SO3·−,从而减少S(IV)的消耗[16, 27-30],提高S(IV)的利用效率。因此,采用FTIR对反应前后的材料表面官能团变化进行分析。如图5(a)所示,反应后C-1000材料表面—COOH含量降低,而官能团C=O和C—H的强度有明显提升,同时在890 cm−1附近发现一个新峰,可归因于Cr—O的拉伸振动[31]。该现象表明—COOH可能与CrSO62−配位形成络合物,并作为电子供体参与Cr(VI)还原。通过XPS表征进一步验证该机理,结果如图5(b)、图5(c)所示。反应前催化剂表面—COOH的峰位置为533.78 eV[32],含量为38.04%,反应后—COOH含量减少至26.05%,表面—COOH峰位置向高结合能方向偏移0.3 eV,这表明—COOH给出电子。图5(d)为反应后催化剂的Cr2p图谱。在587.8 eV和578.3 eV处分别检测到Cr2p1/2和Cr2p3/2,经分析可归属于Cr(VI)[33],这表明Cr可能与C-1000成键。
如图5(e)所示,随着循环次数增加,C-1000对DCF降解的促进效果逐渐减弱,在第3个循环中,DCF的去除率降至80%。这表明C-1000的表面—COOH可能被逐渐氧化,导致催化活性下降。
因此,结合FTIR和Cr2p的XPS图谱,初步推测C-1000表面—COOH可能与CrSO62−形成配合物,同时碳材料的优良导电性促进了配合物的内部电子转移,从而加速SO3·−的产生,减少CrSO62−与HCrO4−的副反应。同时—COOH与S(IV)作为电子给体可进一步提高Cr(VI)的氧化效率,更加快速大量的产生SO3·−、SO4·−等活性物种氧化降解有机污染物。
为了验证该机理,分别通过氧化/还原处理增加/减少碳材料表面—COOH含量,以探究—COOH的作用。将过硫酸铵氧化处理后的C-1000记为OC-t,通过控制氧化处理时间来控制碳材料的表面氧化程度,氧化时间越长,C-1000表面—COOH含量越高[18]。经过水热还原处理的记为RC-T,T代表水热还原处理温度,通过控制温度来控制材料表面官能团的还原[19]。当温度为160 ℃时,碳材料表面-OH被超临界水中的羟基质子化H还原成水,C—O—C发生开环生成—OH,但对—COOH影响不大;而当温度升至200 ℃时,—COOH无法维持稳定被还原成C=O,并进一步还原为—OH。
如图6(a)所示,随着氧化处理的时间增加,OC-t对Cr(VI)/S(IV)体系的促进作用也逐渐增强,其中氧化时间为16 h(OC-16)的体系在20 min时DCF去除率可达95%。另一方面,与C-1000相比,RC-160和RC-200对DCF的去除也出现不同程度的抑制。图6(b)为不同碳材料对DCF的吸附效果,C-1000经水热还原处理后对DCF可产生吸附,这可能归因于在还原处理过程中,碳材料表面官能团结构组成发生改变,从而影响其吸附性能[19,34]。如图6(c)所示,将RC-160与RC-200引入Cr(VI)/S(IV)体系后,DCF的去除率分别为100%和90%,考虑到RC-160与RC-200对DCF分别存在约10%与20%的吸附,经计算其促进分别为30%和10%左右,—COOH含量与DCF氧化降解的促进效果呈正相关。该现象表明—COOH可能是促进DCF降解的主要活性位点。图6(d)中的FTIR图谱也进一步验证了该结果。与C-1000相比,OC-16中—COOH峰强明显增加,—OH峰强略有下降;反应完成后,OC-16中—COOH峰强减弱,C=O、C—OH与C—O—C含量显著增加,并且同样在890 cm−1附近出现可归因于Cr—O拉伸振动的新峰[31]。这进一步证明了碳材料表面—COOH可能与CrSO62−形成配合物,并且该配合物在强化Cr(VI)/S(IV)体系中起到关键作用。
为了进一步验证碳材料—COOH含量对Cr(VI)/S(IV)体系强化效果的影响,对碳材料的O1s图谱进行分析。如图7(a)~(d)所示,与C-1000相比,OC-t材料的表面—COOH含量得到显著提升,并随着氧化时间增加而增加。此外,结合图7(e)可知,碳材料表面—COOH含量变化与对应体系DCF降解反应速率常数变化向吻合。对二者进行定量构效关系(Quantitative structure-activity relationship, QSAR)分析,结果如图7(f)所示。DCF降解反应速率常数与碳材料表面—COOH含量之间具有良好的正相关(R2=0.94)。即—COOH的含量越高,可提供的活性位点就越多,越有利于COO—CrSO6络合物的内部电子转移产生自由基[28],进而加快DCF的降解反应速率,提高污染物去除率并减少S(IV)的副反应消耗。
基于以上结果,提出C-1000强化Cr(VI)/S(IV)体系降解DCF可能的机理,如图8所示。在Cr(VI)/S(IV)体系中,HCrO4−与HSO3−反应生成配合物CrSO62−,CrSO62−与HSO3−进一步反应生成中间配合物[CrO2(SO3)2]*2−,该配合物不稳定,经分子间电子转移分解产生Cr(III)和SO3·−。在C-1000/Cr(VI)/S(IV)体系中,DCF的氧化降解效果得到增强。有研究表明,分子结构内含有羧基基团的有机羧酸可与铬离子配位,通过内球电子转移加快Cr(VI)还原与污染物降解[16]。但C-1000/Cr(VI)/DCF对照体系对DCF的氧化降解贡献较低(图2(a)),因此,推断碳材料表面—COOH可能与CrSO62-发生配位,该配合物借助碳材料的导电性通过内部电子转移反应生产SO3·−,C-1000可作为电子给体参与S(IV)还原Cr(VI)的反应(图5(b)~(c)),该反应抑制了中间配合物[CrO2(SO3)2]*2−的生成,可促进Cr(VI)还原并提高SO3·−的产率,提高S(IV)的有效利用率。C-1000/Cr(VI)/S(IV)体系中SO3·−的产量在短时间内迅速提升,进一步引发自由基链式反应产生强氧化性自由基SO4·−和·OH,实现污染物的氧化降解,其中SO4·−为主要活性物种,·OH有部分贡献。反应结束后,C-1000表面—COOH由于部分参与Cr(VI)的还原而被氧化成碳氧单键(图5(a)),OC-16反应前后的FTIR图谱进一步证实了—COOH官能团的变化(图6(d)),这也是C-1000循环过程中活性降低的重要原因。通过调控材料表面—COOH含量,观察OC-t、RC-T对Cr(VI)/S(IV)体系的活化效果,进行相关性分析后发现—COOH含量与碳材料的催化效果呈线性正相关,该现象证明—COOH在C-1000强化Cr(VI)/S(IV)体系过程中起到关键作用。
-
1)以葡萄糖为原料制备的无金属碳材料可有效强化Cr(VI)/S(IV)体系,提高Cr(VI)的氧化效率和S(IV)的有效利用率,在中性条件下促进产生活性氧自由基,增强对双氯芬酸的氧化降解。
2)体系中产生大量SO3·−,并进一步引发链式反应产生强氧化性SO4·−和SO5·−,其中DCF主要由SO4·−氧化降解,·OH有部分贡献。
3)根据反应前后碳材料表面官能团和组分变化可知,C-1000的表面—COOH可与CrSO62−配位,通过电子内部转移促进Cr(VI)活化S(IV)产生SO4·−,从而降解DCF。
葡萄糖衍生碳材料强化Cr(VI)/亚硫酸盐体系降解水体中双氯芬酸效能与机理
Degradation efficiency and mechanism of diclofenac in waterbody by Cr(VI)/sulfite system strengthened by glucose derived carbon materials
-
摘要: 亚硫酸盐(S(IV))常用于水体中Cr(VI)的还原解毒,在此过程中伴随着SOx·−的产生,可实现水体中有机污染物的同步氧化,但效率不高。本研究以葡萄糖为前驱体,通过简单热解制备无金属碳材料(C-1000),探究在中性条件下C-1000对Cr(VI)/S(IV)体系的促进效果和机理。结果表明,30 min内,在C-1000投加量为0.1 g·L−1时可将目标污染物双氯芬酸(diclofenac, DCF)的去除率由63%显著提高至100%,反应速率提升3.4倍。淬灭实验和EPR实验结果证实SO4·−和·OH共同参与污染物的氧化降解过程,其中SO4·−为主要活性物种,·OH次之。FTIR和XPS表征结果表明,C-1000表面—COOH可能与CrSO62−配位形成三元络合物,利用碳材料的优良导电性,促进络合物分子的内部电子转移,加快SO3·−的产生速率,并通过链式反应促进溶液中SO4·−的产生,以此增强对有机物的降解效果。通过氧化还原处理调控材料表面—COOH含量,建立构效关系进一步验证—COOH的CrSO62−配位作用。Abstract: Sulfite (S(IV)) is commonly used for the reduction detoxification of Cr(VI) in waterbody with the production of SOx·−, which can synchronously oxidize the organic pollutants in water, but the efficiency is low. In this study, glucose was used as a precursor to prepare metal-free carbon materials (C-1000) through pyrolysis, and the promotion effect and mechanism of C-1000 on Cr(VI)/S(IV) system under neutral conditions was explored. The results showed that the degradation efficiency of diclofenac (diclofenac, DCF) was significantly increased from 63% to 100% within 30 min when the dosage of C-1000 was 0.1 g·L−1, and the removal rate was 3.4 times higher than Cr(VI)/S(IV) system. Radical scavenging experiments and EPR results confirmed that both SO4·− and ·OH participated the degradation of DCF, and SO4·− was the main reactive oxygen species, ·OH was followed. The results of FTIR and XPS characterization suggested that —COOH on C-1000 surface may coordinate with CrSO62− to form ternary complexes. The excellent electrical conductivity of C-1000 could facilitate the internal electron transfer of the complexes and accelerate the generation of SO3·− followed by the production of SO4·− via chain reaction, thus the organic degradation effect was strengthened. The quantitative structure-activity relationships of the C-1000/Cr(VI)/S(IV) system were established to further verify the complexation between CrSO62− and —COOH by regulating the —COOH content on the surface through the redox treatment.
-
Key words:
- carbon /
- sulfite /
- Cr(VI) /
- diclofenac degradation /
- degradation mechanism
-
-
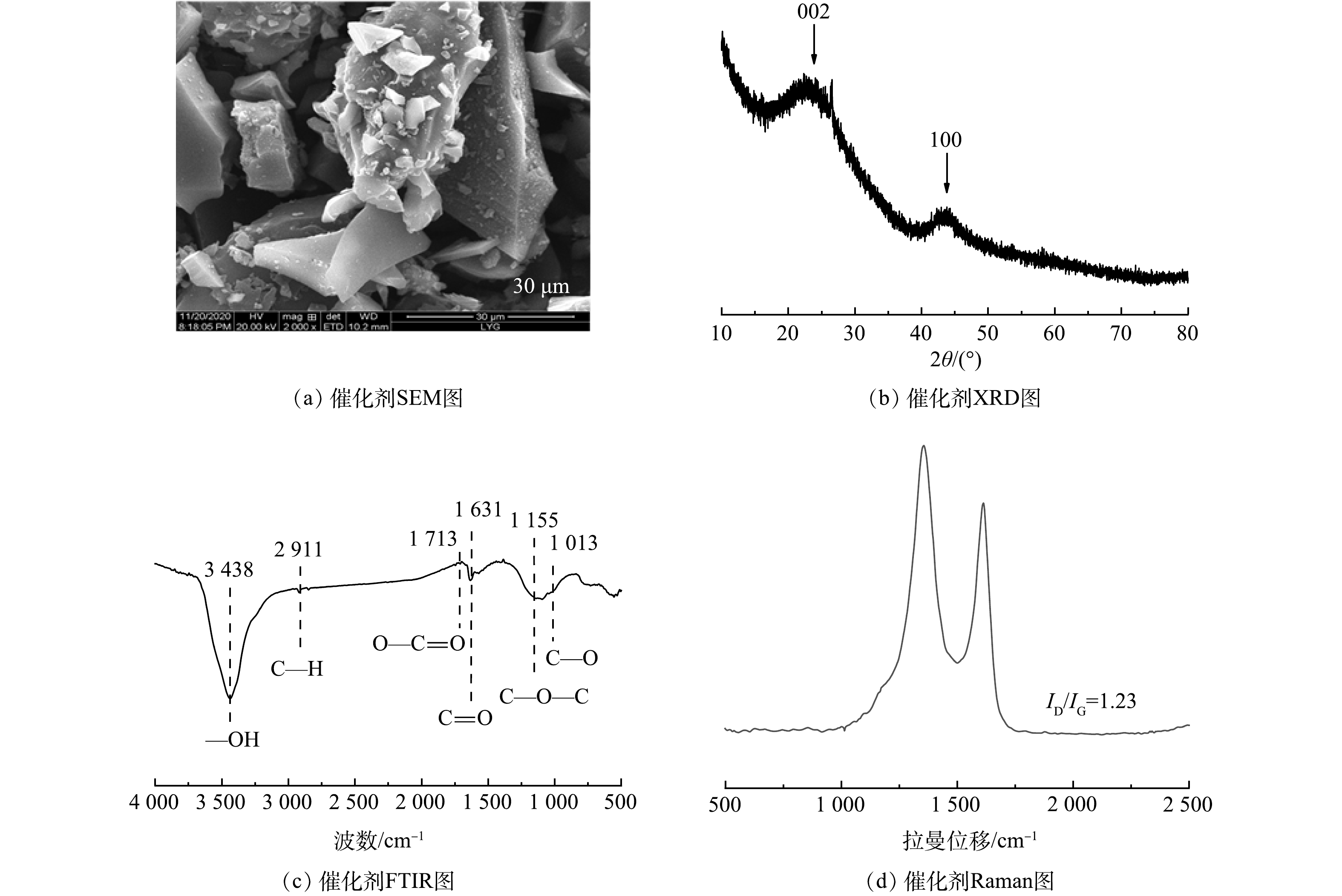
图 1 C-1000的SEM、XRD、FTIR和Raman图谱
Figure 1. SEM image, XRD pattern, FTIR spectrum and Raman spectrum of C-1000
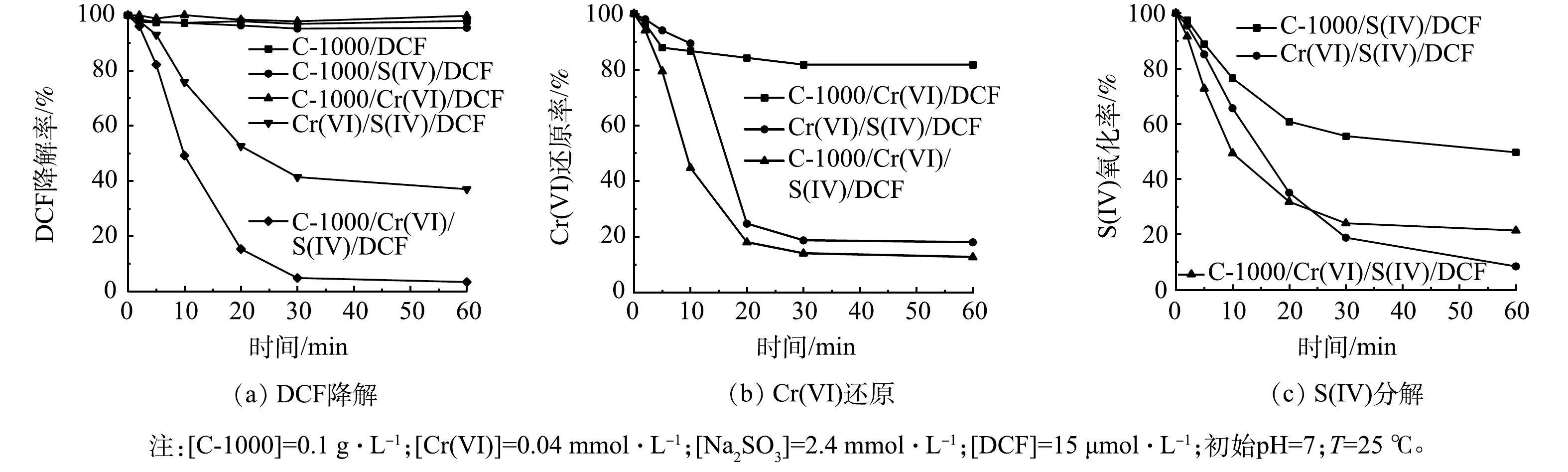
图 2 不同体系下的DCF降解、Cr(VI)还原及S(IV)的氧化曲线
Figure 2. DCF degradation, Cr(VI) reduction and S(IV) decomposition curves in different systems
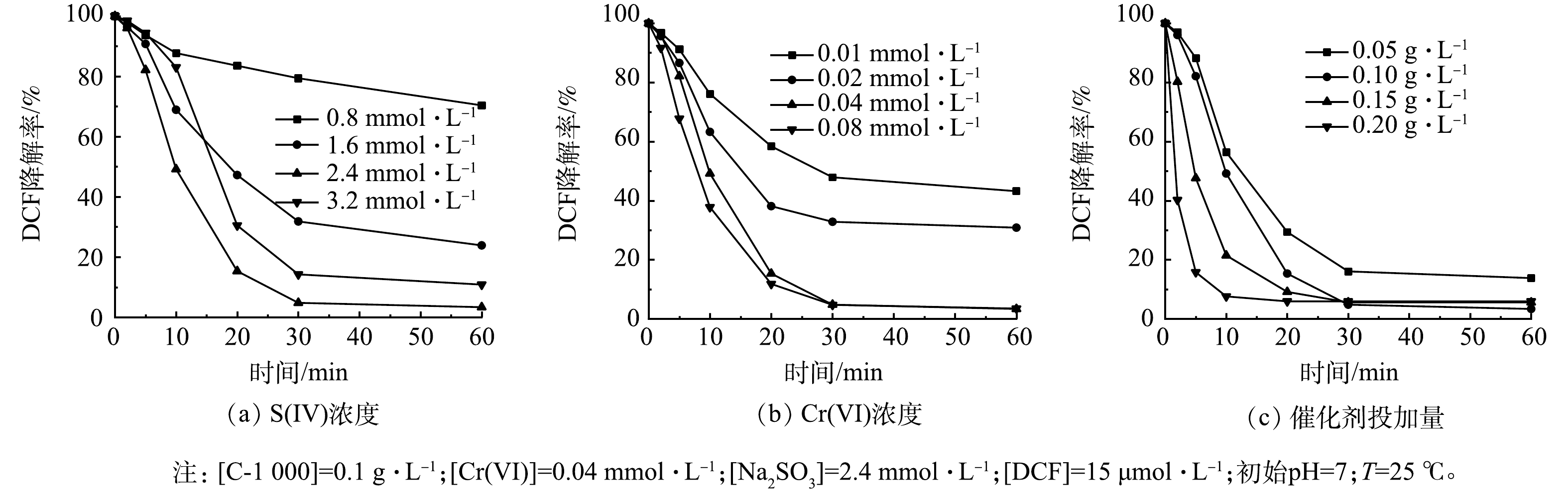
图 3 不同反应条件对DCF降解效率的影响
Figure 3. Effect of different reaction conditions on DCF degradation efficiency
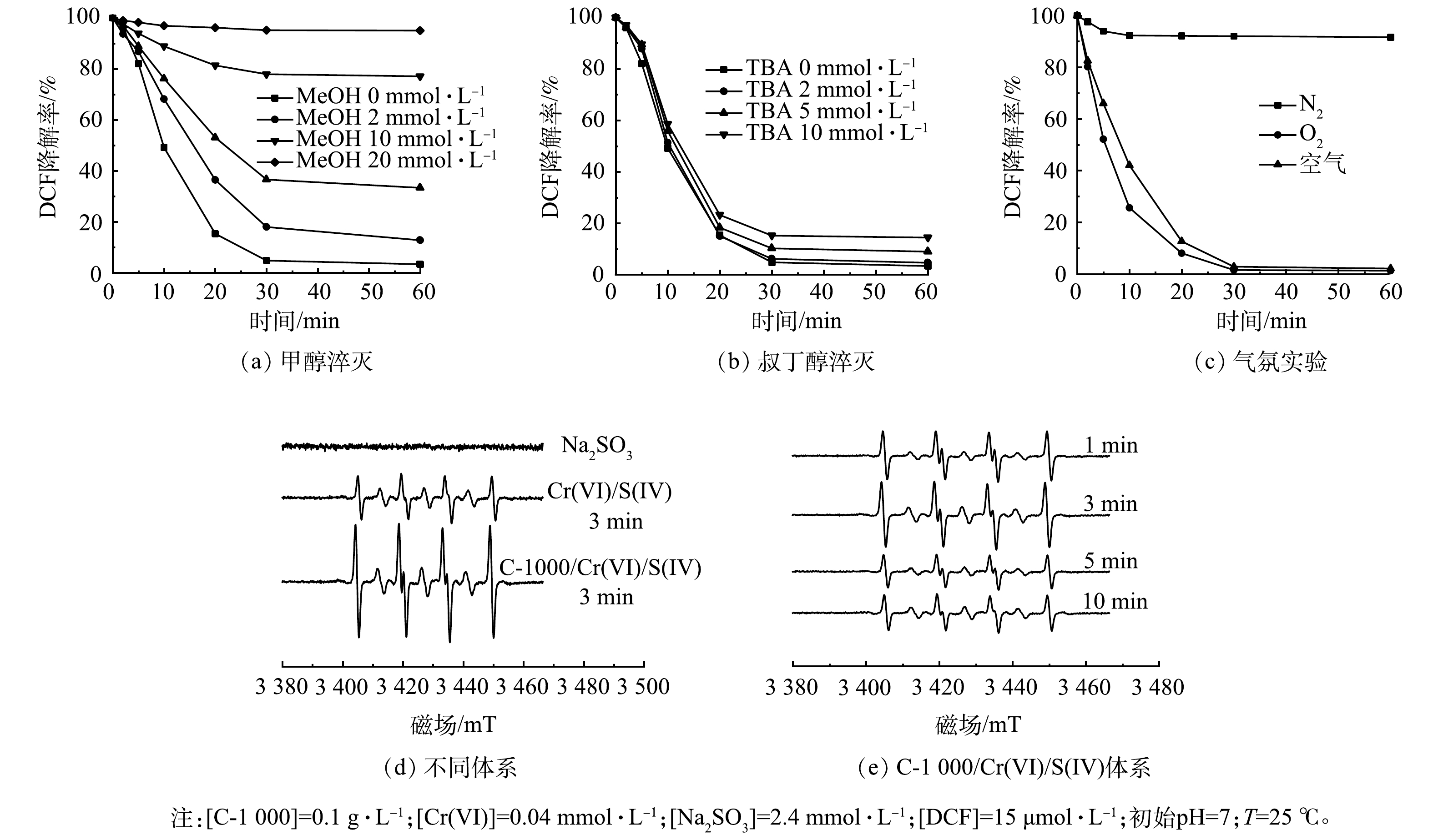
图 4 淬灭实验、气氛实验及不同体系的DMPO的EPR图谱
Figure 4. Quenching experiment, atmosphere experiment and EPR spectra of DMPO obtained in different system.
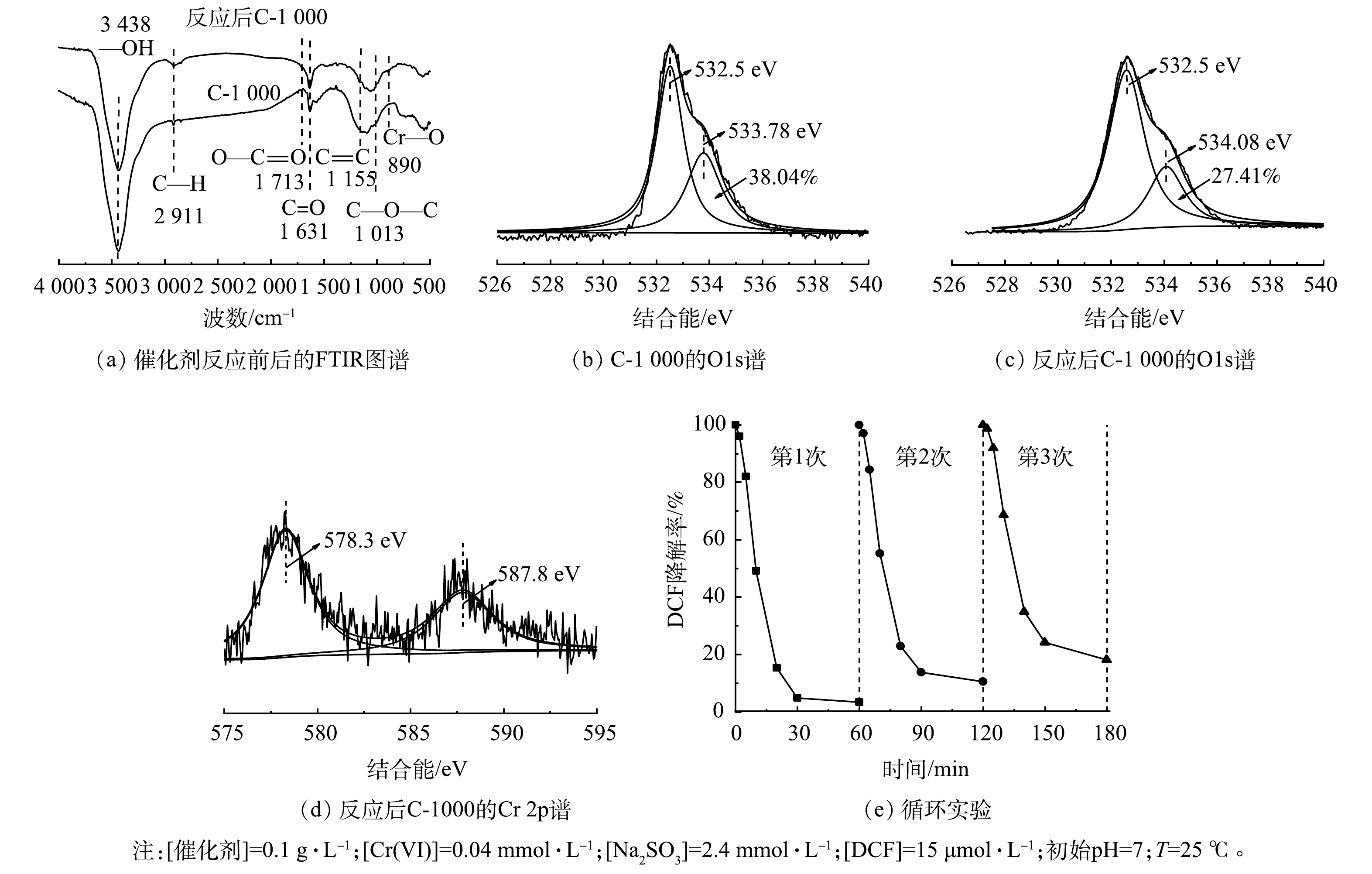
图 5 反应前后C-1000的FTIR图谱、O1s和Cr2pXPS图谱以及循环实验
Figure 5. FTIR spectra, XPS O1s and Cr2p region scan of C-1000 before and after the reaction, and cyclic experiments
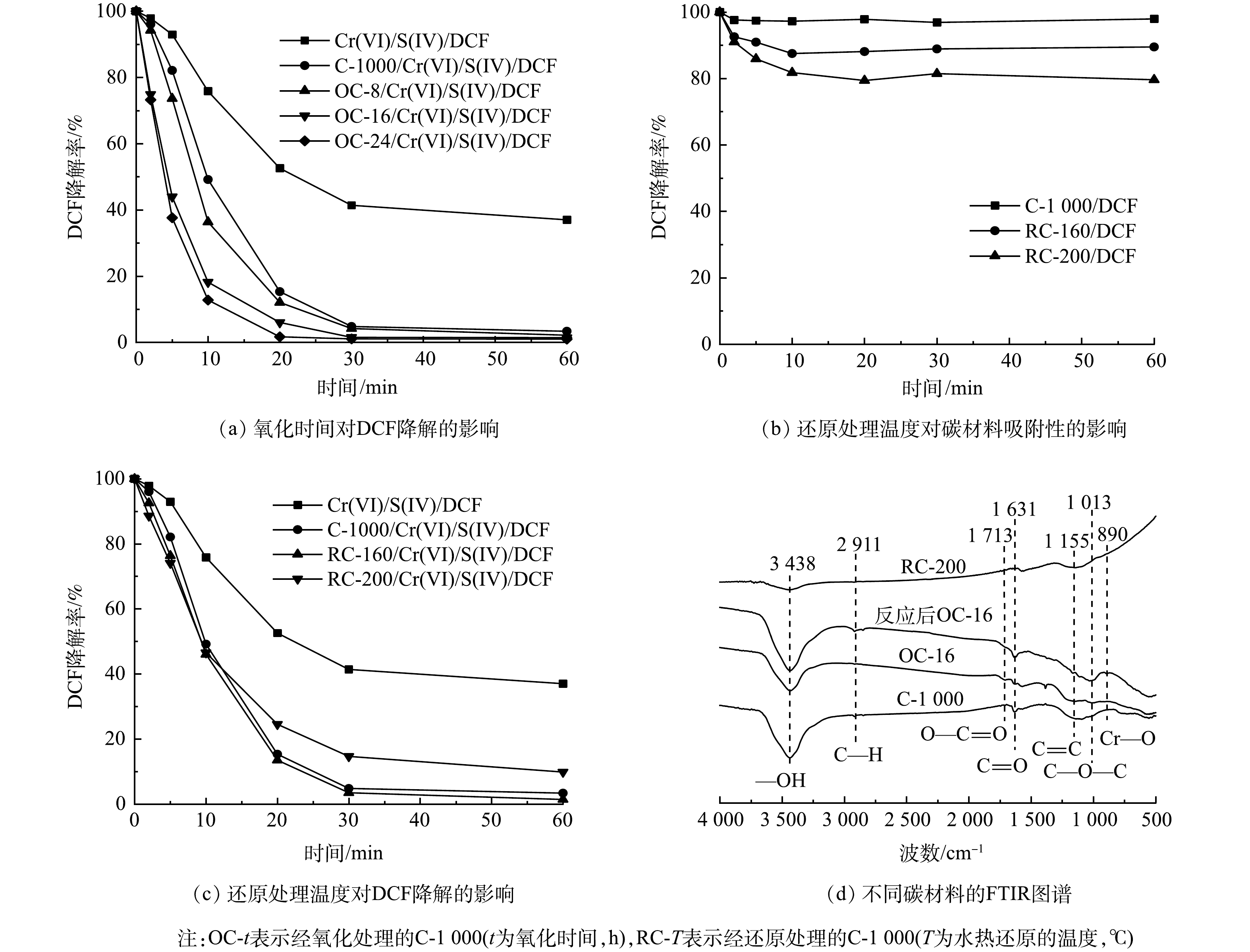
图 6 氧化时间和还原处理温度对Cr(VI)/S(IV)体系吸附和降解DCF效率的影响及不同碳材料的FTIR图谱
Figure 6. Effects of oxidation time and reduction temperature on the adsorption and degradation efficiency of DCF by Cr(VI)/S(IV) systems and FTIR spectra of different carbon materials
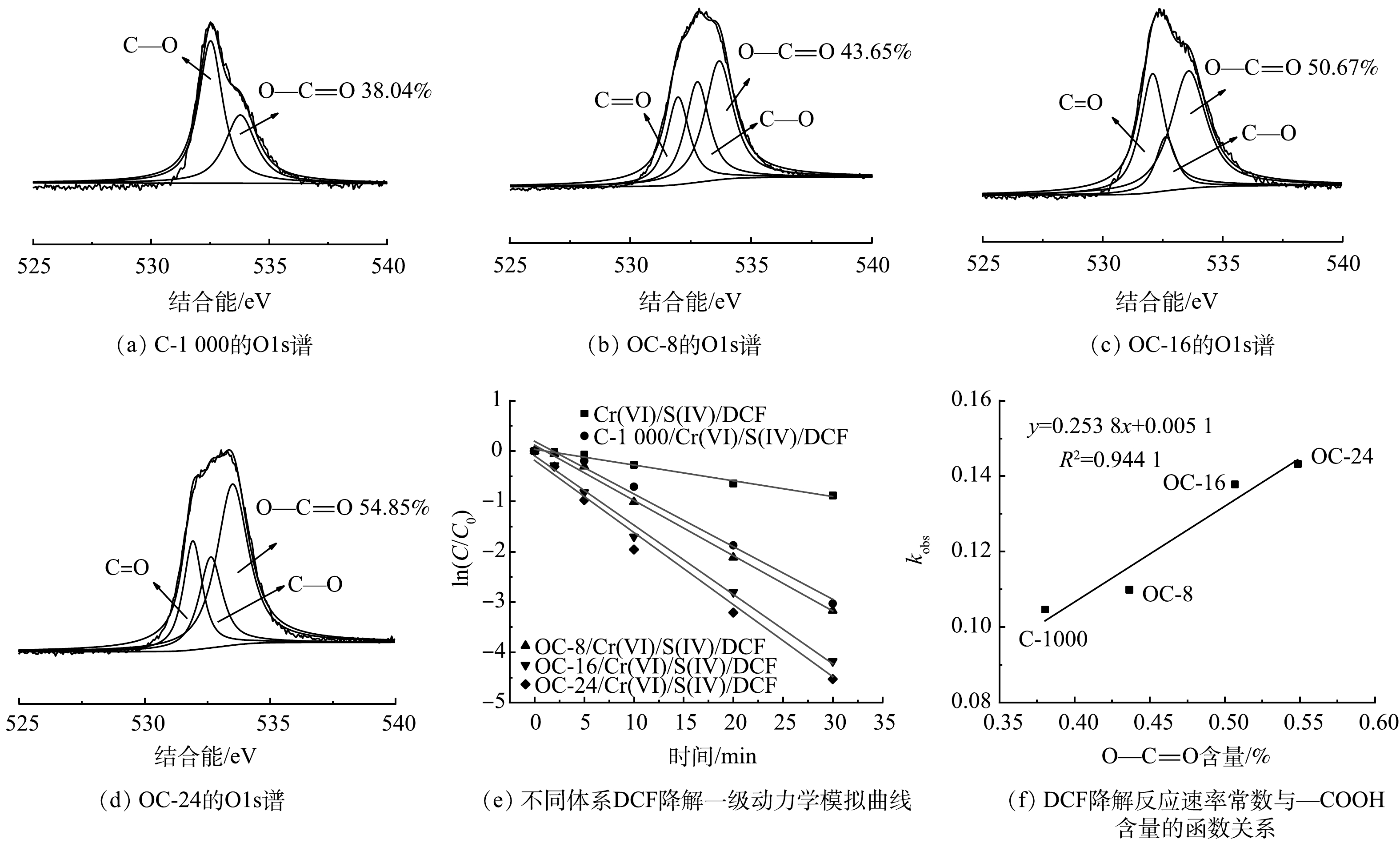
图 7 不同碳材料的O1s谱,不同体系DCF降解一级动力学模拟曲线及DCF降解反应速率常数与—COOH含量的函数关系
Figure 7. O1s region scan of different carbon materials, first-order kinetics model of DCF degradation in different systems, and functional relationship between DCF degradation rate constant and carboxyl content
-
[1] GIL-CARDEZA M L, FERRI A, CORNEJO P, et al. Distribution of chromium species in a Cr-polluted soil: Presence of Cr(III) in glomalin related protein fraction[J]. Science of the Total Environment, 2014, 493: 828-33. doi: 10.1016/j.scitotenv.2014.06.080 [2] CONG Y Q, SHEN L D, WANG B M, et al. Efficient removal of Cr(VI) at alkaline pHs by sulfite/iodide/UV: Mechanism and modeling[J]. Water Research, 2022, 222: 118919. doi: 10.1016/j.watres.2022.118919 [3] DONG H Y, WEI G F, CAO T C, et al. Insights into the oxidation of organic cocontaminants during Cr(VI) reduction by sulfite: The overlooked significance of Cr(V)[J]. Environmental Science & Technology, 2020, 54(2): 1157-1166. [4] HAIGHT G P, PERCHONOCK E, EMMENEGGER F, et al. The mechanism of the oxidation of sulfur(IV) by chromium in acid solution[J]. Journal of the American Chemical Society, 1965, 87: 3835-3840. doi: 10.1021/ja01095a009 [5] BRANDT C, ELDING L I. Role of chromium and vanadium in the atmospheric oxidation of sulfur(IV)[J]. Atmospheric Environment, 1998, 32: 797-800. doi: 10.1016/S1352-2310(97)00331-2 [6] JIANG B, LIU Y K, ZHENG J T, et al. Synergetic transformations of multiple pollutants driven by Cr(VI)-sulfite reactions[J]. Environmental Science & Technology, 2015, 49(20): 12363-71. [7] YUAN Y N, YANG S J, ZHOU D N, et al. A simple Cr(VI)-S(IV)-O2 system for rapid and simultaneous reduction of Cr(VI) and oxidative degradation of organic pollutants[J]. Journal of Hazardous Materials, 2016, 307: 294-301. doi: 10.1016/j.jhazmat.2016.01.012 [8] DONG H Y, WEI G F, FAN W J, et al. Reinvestigating the role of reactive species in the oxidation of organic co-contaminants during Cr(VI) reactions with sulfite[J]. Chemosphere, 2018, 196: 593-597. doi: 10.1016/j.chemosphere.2017.12.194 [9] BUXTON G V, MCGOWAN S, SALMON G A, et al. A study of the spectra and reactivity of oxysulphur-radical anions involved in the chain oxidation of S(IV): A pulse and γ-radiolysis study[J]. Atmospheric Environment, 1996, 30: 2483-2493. doi: 10.1016/1352-2310(95)00473-4 [10] WARNECK P, ZIAJKA J. Reaction mechanism of the iron(III)-catalyzed autoxidation of bisulfite in aqueous solution: Steady state description for benzene as radical scavenger[J]. Berichte der Bunsengesellschaft für physikalische Chemie, 1995, 99: 59-65. [11] ZHANG J M, MA J, SONG H R, et al. Organic contaminants degradation from the S(IV) autoxidation process catalyzed by ferrous-manganous ions: A noticeable Mn(III) oxidation process[J]. Water Research, 2018, 133: 227-235. doi: 10.1016/j.watres.2018.01.039 [12] DAS T N. Reactivity and role of SO5•– radical in aqueous medium chain oxidation of sulfite to sulfate and atmospheric sulfuric acid generation[J]. Journal of Physical Chemistry A, 2001, 105: 9142-9155. doi: 10.1021/jp011255h [13] ZHANG Y, YANG W, ZHANG K K, et al. Sulfite activation by glucose-derived carbon catalysts for As(III) oxidation: The role of ketonic functional groups and conductivity[J]. Environmental Science & Technology, 2021, 55(17): 11961-11969. [14] WANG Z H, MA W H, CHEN C C, et al. Photochemical coupling reactions between Fe(III)/Fe(II), Cr(VI)/Cr(III), and polycarboxylates: Inhibitory effect of Cr species[J]. Environmental Science & Technology, 2008, 42(19): 7260-7266. [15] KRISHNAMURTY K V. , HARRIS G M. The chemistry of the metal oxalato complexes[J]. Chemical Reviews, 1961, 61(3): 213-246. doi: 10.1021/cr60211a001 [16] JIANG B, WANG X L, LIU Y K, et al. The roles of polycarboxylates in Cr(VI)/sulfite reaction system: Involvement of reactive oxygen species and intramolecular electron transfer[J]. Journal of Hazardous Materials, 2016, 304: 457-66. doi: 10.1016/j.jhazmat.2015.11.011 [17] LI N, MA X L, ZHA Q F, et al. Maximizing the number of oxygen-containing functional groups on activated carbon by using ammonium persulfate and improving the temperature-programmed desorption characterization of carbon surface chemistry[J]. Carbon, 2011, 49(15): 5002-5013. doi: 10.1016/j.carbon.2011.07.015 [18] GOSCIANSKA J, OLEJNIK A, NOWAK I, et al. Stability analysis of functionalized mesoporous carbon materials in aqueous solution[J]. Chemical Engineering Journal, 2016, 290: 209-219. doi: 10.1016/j.cej.2016.01.060 [19] GAN G Q, FAN S Y, LI X Y, et al. Effects of oxygen functional groups on electrochemical performance of carbon materials for dechlorination of 1, 2-dichloroethane to ethylene[J]. Chemical Engineering Journal, 2022, 434: 134547. doi: 10.1016/j.cej.2022.134547 [20] REN Y, YUAN Z L, LV K, et al. Selective and metal-free oxidation of biomass-derived 5-hydroxymethylfurfural to 2, 5-diformylfuran over nitrogen-doped carbon materials[J]. Green Chemistry, 2018, 20(21): 4946-4956. doi: 10.1039/C8GC02286K [21] ZHANG K K, SUN P, FAYE M C A S, et al. Characterization of biochar derived from rice husks and its potential in chlorobenzene degradation[J]. Carbon, 2018, 130: 730-740. doi: 10.1016/j.carbon.2018.01.036 [22] ZHANG K K, KHAN A, SUN P, et al. Simultaneous reduction of Cr(VI) and oxidization of organic pollutants by rice husk derived biochar and the interactive influences of coexisting Cr(VI)[J]. Science of the Total Environment, 2020, 706: 135763. doi: 10.1016/j.scitotenv.2019.135763 [23] GU Y, LI H X, LIU L X, et al. Constructing CNTs-based composite membranes for oil/water emulsion separation via radiation-induced “grafting to” strategy[J]. Carbon, 2021, 178: 678-687. doi: 10.1016/j.carbon.2021.03.051 [24] LIU Y J. Simultaneous oxidation of phenol and reduction of Cr(VI) induced by contact glow discharge electrolysis[J]. Journal of Hazardous Materials, 2009, 168(2/3): 992-6. [25] LUO T, PENG Y, CHEN L, et al. Metal-free electro-activated sulfite process for As(III) oxidation in water using graphite electrodes[J]. Environmental Science & Technology, 2020, 54(16): 10261-10269. [26] KHAN A, WANG H B, LIU Y, et al. Highly efficient α-Mn2O3@α-MnO2-500 nanocomposite for peroxymonosulfate activation: comprehensive investigation of manganese oxides[J]. Journal of Materials Chemistry A, 2018, 6(4): 1590-1600. doi: 10.1039/C7TA07942G [27] GHOSH M C, GELERINTER E, GOULD E S. Electron Transfer. 111. Disproportionation of carboxylato-bound chromium(IV). Catalysis by Manganese(II)[J]. American Chemical Society, 1992, 31: 702-705. [28] JIANG B, HE H H, LIU Y J, et al. pH-dependent roles of polycarboxylates in electron transfer between Cr(VI) and weak electron donors[J]. Chemosphere, 2018, 197: 367-374. doi: 10.1016/j.chemosphere.2018.01.047 [29] ZHANG K K, SUN P, ZHANG Y, et al. Enhancement of S(IV)-Cr(VI) reaction in p-nitrophenol degradation using rice husk biochar at neutral conditions[J]. Science of the Total Environment, 2020, 749: 142086. doi: 10.1016/j.scitotenv.2020.142086 [30] HUBER C F, HAIGHT G P. The oxidation of manganese(II) by chromium(VI) in the presence of oxalate ion[J]. Journal of the American Chemical Society, 1976, 98(14): 4128-4131. doi: 10.1021/ja00430a019 [31] JOHNSTON C P, CHRYSOCHOOU M. Mechanisms of chromate adsorption on hematite[J]. Geochimica et Cosmochimica Acta, 2014, 138: 146-157. doi: 10.1016/j.gca.2014.04.030 [32] HU P D, SU H R, CHEN Z Y, et al. Selective degradation of organic pollutants using an efficient metal-free catalyst derived from carbonized polypyrrole via peroxymonosulfate activation[J]. Environmental Science & Technology, 2017, 51(19): 11288-11296. [33] CHOPPALA G, BOLAN N, KUNHIKRISHNAN A, et al. Differential effect of biochar upon reduction-induced mobility and bioavailability of arsenate and chromate[J]. Chemosphere, 2016, 144: 374-81. doi: 10.1016/j.chemosphere.2015.08.043 [34] SUMARAJ, XIONG Z X, SARMAH A K, et al. Acidic surface functional groups control chemisorption of ammonium onto carbon materials in aqueous media[J]. Science of the Total Environment, 2020, 698: 134193. doi: 10.1016/j.scitotenv.2019.134193 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2744
- HTML全文浏览数: 2744
- PDF下载数: 125
- 施引文献: 0
