

-
汞是一种全球污染物,其具有极强的遗传毒性和神经毒性,对人类和生态系统的健康具有广泛影响[1]。大气是汞全球传输的最主要通道,每年全球大约有8千吨汞被排放到大气中,其中人为来源约占39%,自然来源约占6%,以及被二次释放到大气中的汞约占55%左右[2]。大气中汞的存在形式主要有气态元素汞(gaseous elemental mercury, GEM)、活性气态汞(reactive gaseous mercury, RGM)和颗粒态汞(particulate bound mercury, PBM)[3]。其中GEM是大气中汞的主要存在形式,约占大气气态总汞的95%以上[3- 4],并且GEM水溶性较低(3.03×10−7 mol·L−1, 101325 Pa, 25 ℃)[5],化学性质稳定,在大气中的停留时间可长达0.5—2 a,可随大气环流迁移至距离排放源几千公里的地方。因此,在没有明显汞污染源的偏远地区,如南北极、青藏高原等也可检测到显著的人为汞污染[6-7]。RGM为存在于大气气相中氧化态汞化合物,约占大气汞的3 %左右[3],包括HgCl2、HgBr2和Hg(OH)2等多种氧化态汞。RGM化学性质较活泼,在大气中的浓度较低,易溶于水,可吸附在大气颗粒物表面转化为颗粒态汞,因此具有较短的大气停留时间。PBM在大气汞中占比更低,通常低于1%。与RGM类似,PBM可通过干湿沉降从大气中去除。GEM可在太阳辐射、大气氧化物等作用下转化为RGM与PBM,进而沉降至地表;而沉降至地表的汞还可通过化学与生物还原转化为GEM[8-11],再次进入到大气中,从而进行长距离迁移。
大气中汞的氧化还原反应可改变其化学形态,对汞的环境行为具有至关重要的作用。例如,在极地地区春季大气汞“亏损”事件中[12],溴自由基引发零价汞的氧化导致大气零价汞显著降低[13],氧化态汞的沉降也使极地地区成为汞重要的汇[14]。此外,大气中存在的多种氧化剂如O3、·OH、NO3和各种卤素自由基等,可将GEM氧化为RGM。同时,RGM又可以通过光致还原反应转化为GEM[9, 15]。大气中RGM的液相还原主要发生在大气液滴中,由于Hg0的较高蒸汽压与低的汞溶解性,液相还原生成的Hg0可快速扩散至气相,导致大气中Hg0的浓度增加和半衰期延长。但是大气水相的均相和非均相研究主要以实验和地学模型为主,不是本文讨论的重点,不在此进行详细论述。大气中零价汞(Hg0)与一价汞(HgⅠ)和二价汞(HgⅡ)的主要光化学转化机制如图1所示。为评估全球汞循环,建立汞全球传输模型,需要深入了解大气汞的氧化还原过程。但是大气中自由基与气态元素汞的含量均极低,且氧化还原反应中过渡态存在时间极短,给大气汞氧化还原机制的实验研究带来了诸多困难。同时,大气反应的实验室模拟通常存在器壁效应[16],与真实大气反应存在一定的差异。随着计算机技术的快速发展,计算化学越来越多地应用于大气汞的化学转化机制研究。本文从计算化学角度,总结了近年来GEM被大气氧化剂氧化的理论研究进展,包括氧化产物的稳定性研究、反应机制的理论推测以及反应速率的计算等;同时还总结了采用高精度量子化学方法计算大气中HgⅠ和HgⅡ直接进行光解反应的反应分支比和反应速率等最新研究进展。
-
自极地大气汞“亏损”事件[12]发现以来,大气汞的氧化反应受到了越来越多的关注,研究人员对GEM的氧化过程展开了一系列研究。大气中的氧化剂包括O3、·OH、NO3和各种卤素自由基等均可能将GEM氧化为RGM,但是其氧化机制和反应速率在不同环境条件下仍存在极大的不确定性。量子化学家们采用各种量子化学理论和方法,开展了计算模拟研究大气元素汞氧化反应的热力学参数、动力学参数以及微观反应过程,从微观角度深入了解和探索了大气汞的氧化机制。
-
臭氧(O3)是大气中重要的气相氧化剂。早期研究发现,极地大气Hg0浓度与对流层中O3浓度呈正相关,进而推测O3是大气Hg0的主要氧化剂。反应(1)被认为是大气Hg0的主要氧化途径[17]:
其中,Hg0与O3反应的中间体可能是HgO3[17],HgO被认为是最终产物。产物的稳定性对于反应的正向进行起至关重要的作用。键解离能可以直接用以判断化合物的稳定性,键解离能越高,化学键越强,分子越稳定。采用非迭代三重激发模式的耦合簇方法(coupled-cluster single, double, and triple excitation method, CCSD(T))和多参考组态相互作用(multi-reference configuration interaction, MRCI)计算HgO在0 K下的键解离能[18]为16.7 kJ·mol−1,表明HgO是极弱的键,Hg与O之间是范德华作用,极易分解。然而,实验测得HgO的键解离能[19]为(221.8±41.8)kJ·mol−1,与计算相去甚远。一般而言,计算与实验值相差为±4.2 kJ·mol−1左右。研究[19]认为HgO计算的键解离能与实验值相差如此之大的原因在于,实验测定的是HgO的二聚体,或者是单体与二聚体和三聚体的混合物,而非HgO单体。当HgO形成二聚体(HgO)2,未配对的电子几乎均定域在氧原子上,同时Hg−Hg之间形成了π键,稳定了分子,因此HgO分子更易于形成二聚体。该研究计算了HgO二聚体如O·Hg2·O和Hg·O2·Hg的键解离能分别为523 kJ·mol−1和536 kJ·mol−1,当折算成每个单体的键解离能时,接近实验测定[19]的(221.8±41.8) kJ·mol−1。
反应的热力学参数是判断反应进行方向的重要依据。1995年曾有实验[20]测定298 K下反应(1)的吉布斯自由能为−171 kJ·mol−1,当HgO为固态时,反应的吉布斯自由能为−254 kJ·mol−1,从热力学角度表明反应(1)可以自发进行。但是,当采用高精度量子化学方法CCSD(T)/CCD计算了在298.15 K下气态HgO的生成吉布斯自由能[21]为51.8 kJ·mol−1,表明产物HgO是一种不稳定的化合物;同时计算出反应吉布斯自由能为69.5 kJ·mol−1,表明该反应并不是自发进行的。实验与计算产生差异的原因可能是实验中气相HgO会进一步与H2O反应生成Hg(OH)2,该反应为强放热反应[17]。另外,实验测定的反应产物可能为HgO的二聚体或者单体和多聚体的混合物[19],这也可能导致实验与计算产生差异。O3氧化Hg0的反应机制尚存较多不确定性,有研究[17]推测在真实大气环境中Hg0与O3反应生成中间体OHgOO,然后分解为HgO和O2后,HgO会继续分解为Hg和O,然后O与O2反应又生成O3;而在实验条件下中间体OHgOO会与H2O或者器壁界面碰撞而生成固体HgO(s)和O2,然后与H2O继续反应生成Hg(OH)2;在真实大气环境中,只有当产物HgO遇到气溶胶或者接触海洋表面时,HOHgOH才可能转化为各种二价汞离子,否则可能仍然以Hg0存在。
热力学参数是判断反应进行方向和产物稳定性的重要依据,反应动力学参数可以判断反应的快慢,反应速率常数的计算通常需要明确反应路径。由于O3氧化Hg0的机制尚不明确,该过程的速率常数研究仍以实验测定为主。实验测定反应(1)的速率常数在4.9×10−18 — (3±2)×10−20 cm−3·molecule−1·s−1之间[20, 22-24],根据测定的反应速率计算Hg0在大气中的半衰期约为10.6天[25],这显然与Hg0在大气中的实际半衰期不符。实验测定速率计算得到半衰期与GEM在大气中实际半衰期不一致的原因可能与实验室模拟反应存在器壁效应,以及实验室模拟反应与真实大气中GEM发生的反应不完全一致有关。
-
羟基自由基(·OH)是大气中常见的重要氧化剂,最初[17, 26]认为其对Hg0氧化起重要作用,主要通过反应(2)氧化Hg0:
其中,M为除·OH和Hg0以外的第三种物质。Hg—OH键能大小与其稳定性直接相关。早期采用较低水平的计算方法B3LYP计算了Hg—OH的键解离能在39.4—39.7 kJ·mol−1 左右[27-28]。随着计算机技术的飞速发展,最近研究采用耦合簇方法CCSD(T)/AVTZ计算了Hg—OH的键解离能[29-30]为46.1—51.9 kJ·mol−1。一般认为化学键的键能在41.8 kJ·mol−1左右,为极弱键,易于分解。以上计算表明,·OH即使可将Hg0氧化为HgOH,也会快速分解回Hg0和·OH,不利于反应(2)正向进行。也有研究认为HgOH可进一步与大气中的O2快速发生以下反应(3)[29]:
但是计算反应(3)的反应焓变[25]约为210 kJ·mol−1,为吸热反应;同时HgO的键解离能[18]较小,约为16.7 kJ·mol−1,易于分解为Hg和O。虽然HgOH与大气中的O2不易继续发生反应,但是如果遇到大气中其他高浓度的氧化剂Y,HgOH可以与Y继续反应生成二价汞,如反应(4)所示[17]。这一反应会与HgOH的解离反应发生竞争,从而稳定HgOH,有利于·OH氧化Hg0最终转化为HgII。
采用高精度量子化学计算方法CCSD计算产物HOHg—Y的键长和键解离能[29],结果显示当氧化剂为O2时,键解离能最小(D0 = 38.9 kJ·mol−1);氧化剂为OBr·时,键解离能最大(D0 = 237.2 kJ·mol−1)。这一计算结果表明,HgOH可能与大气中的其他氧化剂进一步生成稳定的RGM,但这也取决于大气中该氧化剂的浓度。当Y的浓度太低时,HgOH解离仍占主导。
羟基自由基氧化Hg0反应速率现多为实验模拟测定值,尚无基于反应(1)的能垒计算反应速率的报道。采用相对速率实验方法测定了Hg0与·OH在295 K和1个大气压下的气相反应动力学,得到反应速率常数[26]为(8.7±3)×10−14 cm3·molecule−1·s−1;通过激光诱导荧光检测当Hg0过量时·OH的浓度,测定出反应(2)的速率上限[31]为1.2×10−13 cm3·molecule−1·s−1 (298 K、53328 Pa)。将·OH氧化Hg0的数据代入汞的GEOS全球化学与迁移模型,结果显示,对于少量高污染的地区,·OH氧化Hg0所占HgⅡ比例可高达83%,但这一氧化过程对全球范围大部分地区HgⅡ来源贡献小于1%[29]。基于此,目前研究者普遍认为·OH氧化Hg0的反应对于大气中Hg0的转化作用不大[2, 27, 29]。
-
溴原子引发的Hg0氧化在春季海洋边界层大气汞“亏损”事件[12, 13]中起重要作用。溴原子首先通过反应(5)将Hg0氧化为BrHg·,其中BrHg·也可解离回Hg0,或者继续与大气中氧化剂进一步发生反应(6)[13, 27, 32-33]。
采用密度泛函B3LYP/CEP-121G方法计算的0 K下BrHg·键解离能[27]为63.8 kJ·mol−1;采用耦合簇方法CCSD(T)/CBS+CV+SO计算出0 K下BrHg·键解离能[34]为68.3 kJ·mol−1,计算结果和实验测定[35]的(65.0±1.2)kJ·mol−1相近;以上结果表明BrHg·相对于HOHg·和HgO更稳定。采用高精度计算化学方法CCSD(T)/CCD获得反应(5)的吉布斯自由能[21]为−33.5 kJ·mol−1,属于放热反应;因此相较于·OH和O3,Br·氧化Hg0在热力学上更有优势。
虽然BrHg·的键能较HgOH和HgO更大,但BrHg·依然属于不稳定化合物。目前尚无实验证实BrHg·的存在。当BrHg·与大气中的氧化剂继续发生反应(6)时,BrHg−Y的稳定性决定了反应进行的方向。表1给出了不同方法计算的BrHg—Y的键解离能。从表1中可以看出,BrHg—OBr和BrHg—OCl的键解离能[36]最大,分别为232.6 kJ·mol−1和220.6 kJ·mol−1;BrHg—OO和BrHg—NO的键解离能[36-37]最小,分别为27.9 kJ·mol−1和49.2 kJ·mol−1,热力学上认为其不能稳定存在;当Y为·NO2和·OOH时,BrHg—Y也较稳定,键解离能均大于100 kJ·mol−1。有研究报道HgBrONO和HgBrOOH是Br引发的Hg0氧化的主要氧化产物[32, 36, 38],其中syn-HgBrONO则是热力学最稳定的产物。尤其在极地积雪间隙的空气中,·NO2对BrHg·的氧化起重要作用[39]。同时,通过高精度计算化学PBE0和CCSD(T)方法计算BrHg·与·NO2反应路径和能垒[40],计算结果显示syn-BrHgONO是BrHg·与·NO2反应的最稳定产物,相对于BrHgNO2和anti-BrHgONO,syn-BrHgONO的能量最低,分子最稳定。而计算研究[40]发现BrHg·+·OOH→BrHgOOH相对于BrHg·+·OOH→BrHgH+O2和BrHg· + ·OOH→Hg+BrOOH,不仅没有反应能垒,而且所得产物BrHgOOH能量最低,最稳定。具体环境中,BrHg·被·NO2和HO2·氧化的比例主要依赖于环境中·NO2和HO2·的浓度大小。大部分的Hg0都是在对流层被氧化为HgⅡ,并且北半球比南半球更快,这主要是因为北半球溴原子与·NO2的浓度更高[32]。在2017年的GEOS-Chem模型研究中也指出,BrHg·大部分是被·NO2氧化为HgⅡ,其次是HO2·,其他·ClO,·BrO和·OH氧化剂所占比例远小于·NO2和HO2·,这与对流层上部这些氧化剂的浓度有关[32]。同时,动力学研究发现[32, 39]只有·NO2和HO2·与BrHg·生成HgⅡ的速率大于BrHg·的解离速度。除此之外,由于计算获得Hg—H和Hg—C的键能太小,BrHg·几乎不会从大气中的微量挥发性有机物中提取H,也不会与含不饱和双键的乙烯类气体发生加成反应[36, 38]。
Br原子氧化Hg0的反应速率既可通过实验室模拟大气反应获得,也有理论计算报道。采用不同实验方法测定反应(5)的速率常数为(1.6±0.8) ×10−12—3.6×10−12 cm3·molecule−1·s−1[16, 41-42]。采用RRKM(Rice-Ramsberger-Kassel-Marcus)理论和从头算方法计算298 K、101325 Pa的Hg0和Br原子速率常数[27]为1.1×10−12 cm3·molecule−1·s−1 ;用变分过渡态理论(Variational transition state theory, VTST)计算了298 K、101325 Pa的速率常数[43]为2.04×10−12 cm3·molecule−1·s−1;采用耦合簇方法CCSD(T)和准经典轨迹方法(quasiclasical trajectories,QCT)计算其在260 K、105 Pa下的速率常数[44]为1.2×10−12 cm3·molecule−1·s−1。从以上结果可以看出,计算与实验测定的速率常数均在同一数量级;相较于·OH、O3等与Hg0的反应速率,Br氧化Hg0的速率明显更大。对于反应(6)的反应速率,目前研究的还较少,有研究采用高精度量子化学方法CCSD(T)计算了BrHg·与·NO2和HOO·的反应速率常数[40]。
氯原子和溴原子同为卤素,有研究认为它也可引发Hg0的氧化[36];其反应机制与溴原子类似,也是分两步将Hg0氧化为HgⅡ:首先生成ClHg·中间体,然后与大气中的其他氧化剂(Y=Cl·、·NO、·NO2、O2、HO2·、·BrO、·ClO)反应生成ClHgY。ClHg−Y的键解离能如表1。从表1可见,ClHg—Y和BrBr—Y的键解离能类似,ClHg—OBr和ClHg—OCl键能最大,分别为237.0 kJ·mol−1和225.1 kJ·mol−1;其次是ClHg—OOH、ClHg—ONO和ClHg—NO2,分别为183.1、165.0、153.4 kJ·mol−1;ClHg—NO和ClHg—OO键能最弱,分别为54.4 kJ·mol−1和33.0 kJ·mol−1,其在热力学上是不稳定的。采用相对反应速率[42],测定了Cl原子和Br原子与Hg0氧化反应的速率分别为(1.8±0.5)×10−11 cm3 · molecule−1·s−1和(1.6±0.8)×10−12 cm3·molecule−1·s−1;其他研究测定了Cl原子与Hg0的反应速率[16]为(1.0±0.2)×10−11 cm3·molecule−1·s−1。从反应速率可以看出,Cl原子与Hg0的反应比Br原子稍快,动力学上更有利。
除此之外,实验观察到·BrO可以将Hg0氧化为HgⅠ和HgⅡ,通过质谱鉴定出产物HgOBr/HgBrO、HgBr、HgO[45]。研究指出·BrO有可能引发Hg0的氧化反应,生成BrHgO[46]。但是该反应要求一个未激发的Hg0原子插入到一个强键Br—O之间,这在理论上很难发生;Br—O的键能为238 kJ·mol−1左右,该反应的能垒为150 kJ·mol−1,这在动力学上也很难进行。也有研究[47]认为·BrO与Hg0的最终产物为Hg—BrO,生成的Hg—BrO键能约为80 kJ·mol−1,在理论上更可行。而Hg0与·BrO反应生成Hg—OBr,其键能更低,计算值甚至是负值;Hg—OCl键能[36]也仅为2.2 kJ·mol−1。因此,理论上HgOBr、HgOCl是极不稳定的化合物,不能稳定存在。
-
虽然目前大部分研究工作聚焦于臭氧、羟基自由基和卤素引发的Hg0氧化,仍有研究者提出Hg0可能存在一些其他氧化机制,如H2O2和NO3对Hg0的氧化。有研究[49]在实验室模拟了H2O2和CH3I与Hg0的氧化反应,测定其反应速率分别为5.6×10−19 cm3·molecule−1·s−1和(0.3—15)×10−21 cm3·molecule−1·s−1;而实验室模拟NO3氧化Hg0的反应[5, 50],测定的反应速率为4×10−15 cm3·molecule−1·s−1,但实验并未测得推测的反应产物(HgO和NO2)。另一方面,采用高精度量子化学计算HgO的键解离能仅为16.7 kJ·mol−1左右[18],表明HgO并不是一种稳定的化合物,HO2和NO3均不能与Hg0形成稳定的氧化物,因此不能引发Hg0的氧化[36]。虽然最近的一项研究[51]指出,夜间NO3的浓度与HgⅡ呈现正相关性(R2=0.47),因此推测NO3可能参与了HgⅠ的氧化;但该研究并未对HgⅡ与其他环境变量的相关性进行详细研究,目前并没有充足数据证实其参与到Hg0的氧化。
-
GEM不仅可以在气相中被氧化,也可能在大气液相和颗粒物上发生氧化反应,但是具体过程和机制仍不清晰。已有研究发现臭氧和羟基自由基可以将大气气溶胶中的GEM氧化为RGM[52];同时实验测定大气中颗粒物的浓度与颗粒态汞的浓度正相关,这可能与颗粒物对GEM的吸附和氧化有关[53]。另外,采用非质量同位素分馏方法研究发现大气降雨中偶数汞同位素的非质量分馏可能与对流层气溶胶或者固体表面发生的GEM氧化有关[54]。在实验条件下,用铁氧化物纳米颗粒代替大气气溶胶矿物组分模拟其对GEM的吸收,发现这一过程可能涉及了GEM的氧化,但是具体过程和机制还需进一步研究[55]。目前,GEM在大气中发生的非均相氧化反应多以实验测定为主,量子化学计算目前研究较少。另外,化石燃料的燃烧、金属冶炼和水泥生产等也是大气中汞重要的释放来源[2, 56-57],其中煤炭的燃烧是最主要的人为汞排放来源之一。在煤高温燃烧时,煤中的汞绝大部分以Hg0存在[58-59];随着烟气的冷却,部分Hg0与烟气中卤素、酸性气体和氧气等发生均相反应生成Hg2+ [59-61];随着温度的继续降低,Hg0会在飞灰和烟气组分的作用下,发生非均相反应生成Hg2+ [58,62-64],或者通过飞灰吸附形成颗粒态汞。整个燃烧过程影响汞形态转化的因素很多,例如煤的种类、烟气组分、燃烧温度、空气污染控制设备的种类和实际运行工况等,该过程研究不是本文讨论的重点。
-
目前人们已经普遍意识到GEM氧化反应的重要性,并且将部分氧化反应添加到大气传输模型中,但对大气中RGM还原反应的研究仍然极为匮乏。早期研究认为RGM的还原反应仅存在于大气水相如云层或者气溶胶[32],但是最新计算研究显示HgⅡ化合物[65](如syn-HgBrONO、anti-HgBrONO、HgBrOOH、HgBrOH、HgBrNO2、HgBr2、HgCl2、HgBrOCl、HgBrI、HgBrOBr、HgBrOI)和HgⅠ化合物[15](如HgBr、HgCl、HgOH、HgI)也可吸收紫外可见光直接发生还原反应。因此,RGM可以在日间吸收太阳光发生光还原,这可能是大气中HgⅡ和HgⅠ化合物的主要还原途径。同时,相比于之前将RGM的还原反应局限在大气水相,这一新的还原机制的提出可能会进一步缩短RGM在大气中的停留时间。目前已经在青藏高原[66]和城市空气[67]中观测到氧化态汞的光还原现象。同时,有实验证实了syn-HgBrONO可发生快速光解还原反应生成HgBrO自由基,该自由基会继续与大气中的挥发性有机物发生反应生成HgBrOH[68]。以下将从直接光解还原和间接还原两个方面介绍RGM在大气气相中的还原反应机制。
-
在光照条件下,RGM吸收光能量从基态(S0)跃迁到激发单重态(Si),激发态再裂解或转化为光解产物。其中,对于HgⅡ和HgⅠ化合物的直接光解作用,激发态的研究是光解反应的难点与重点。对于HgⅠ化合物,早期计算研究了HgX(X为卤素)自由基发生B→X转化/跃迁的寿命[69]、HgCl和HgBr的低电子态以及发生X−A转化/跃迁的两种电子态[70-72]。最近,有研究采用CASSCF方法以及MRCI方法计算了HgⅠ自由基如HgBr、HgCl、HgI和HgOH不同电子态的势能曲线、键长、键角以及键解离能,并用MS-CASPT2方法计算预测了这些HgⅠ自由基的吸收光谱和吸收截面[15]。该研究表明大气中的HgⅠ自由基发生光还原作用将与Hg0的氧化作用竞争,同时该机制将会影响汞在大气中停留时间的估算。
对于HgⅡ化合物,已有研究显示大气中Br原子氧化Hg0的HgⅡ产物主要为syn-HgBrONO和HgBrOOH[32, 36, 38]。早期主要对闭壳层分子的热力学性质进行研究[73-75],最近研究采用非绝热动力学定量计算了HgⅡ的化合物如syn-HgBrONO、HgBrOOH、HgBrOH和HgBrO直接光解的反应热力学和动力学,其光解反应如(7)—(16)所示[9]。同时,已有研究采用实验方法证实syn-HgBrONO发生快速光解后生成HgBrO自由基,并继续与大气中挥发性有机物发生反应生成HgBrOH[68]。
syn-HgBrONO中的电子从n3(ONO)轨道跃迁到π*(ONO)轨道形成S1激发态,然后发生直接光解其反应如(7)和(8)所示,其中90%的产物为HgBrO和NO,10%的产物为HgBr和ONO[9],该计算结果也与报道syn-HgBrONO的主要光解产物为HgBrO一致[68]。HgBrOOH和HgBrOH有3种主要的光解反应通路,如(9)—(11)和(12)—(14)所示。其中Hg—Br、Hg—O和O—O键会发生解离,反应(9)和(12)分别是HgBrOOH和HgBrOH的主要光解反应[9]。HgBrO是syn-HgBrONO和HgBrOOH的主要光解反应中间体,其可能发生进一步光解生成Hg0,也可能与大气中的NO、NO2等发生反应生成HgBrONO、HgBrONO2和HgBrOH[9]。HgBrO的光解反应通路如(15)和(16)所示,其中反应(15)为主要的反应通路,HgBrO吸收300—500 nm波长的太阳光,基态D1中O原子2p轨道上电子跃迁到Br原子4p轨道上形成D4和D5激发态,然后发生光解离反应生成HgⅠ或者Hg0。这与最近报道的大气中HgⅠ化合物如HgBr、HgCl、HgI和HgOH会吸收太阳光发生光解反应的结论一致[15]。
光化学反应的反应速率与反应物的浓度无关,而是与照射的光强度有关,其计算公式[15]为:
近年来,Saiz-Lopez等采用多参考完全活性空间-二级微扰理论(multireference complete-active-space self-consistent field/multistate completeactive-space second-order perturbation theory (CASSCF/MS-CASPT2) method)方法计算了HgⅡ的化合物如syn-HgBrONO、HgBrOOH、HgBrOH和HgBrO直接光解的速率常数,总结在表2中。除此之外,早期有研究采用公式 k−1(T)=Keq(T)/k1(T)来计算了HgBr的热分解速率,即根据反应(5)的平衡常数[27]除以HgBr的生成速率常数[41]得到HgBr的解离速率常数,为1.6×10−9 cm3·molecule−1·s−1获得,其数据如表2所示。
-
RGM也可与大气中的还原剂作用,发生间接还原反应。最新研究[8]采用高精度计算化学方法(MP2+CCSD(T)/aug-cc-pwCVnZ),计算出BrHgO·与CO可以发生还原反应生成BrHg·和CO2,其中反应焓变为−282 kJ·mol−1,由于产物BrHg·的键解离能仅为65 kJ·mol−1,小于BrHgO·和CO反应的焓变,则BrHg·可进一步分解为Br和Hg0;并根据反应能垒和阿伦尼乌斯公式计算了200 K和298 K下的反应速率常数为2.9 ×10−11 — 9.4×10−12 cm3·molecule−1·s−1,该反应机制的提出进一步丰富了RGM在大气中可能发生的还原反应通路。另外,早期有研究[76]指出大气中的HgO也可被CO还原生成Hg0,反应速率为5×10−18 cm3·molecule−1·s−1;此外,在燃煤电厂烟羽中的SO2也可能导致RGM发生气相还原[77],但是目前采用量子化学计算方法对这些反应的机理进行报道的较少。
-
汞作为一种全球污染物,大气中元素汞和氧化态汞的化学转化对全球汞的沉降和迁移距离具有决定作用。虽然目前已经提出了多种大气汞的氧化还原机制,但依然不能完全解释各种形态的汞其时空变化。最新研究显示[8],当将主要的汞氧化还原机制的反应速率代入到全球汞化学传输模型(GLEMOS),RGM的光致还原反应很大程度上抵消了GEM的氧化过程,导致计算出的大气汞停留时间远高于实验测定值,也表明现有汞传输模型中依然缺乏汞氧化机制。量子化学计算方法从微观角度研究反应机制,可获得热力学参数、动力学参数以及反应轨迹等信息,为研究大气汞的迁移转化提供了巨大助力。但是由于大气中各种形态汞浓度极低,反应中间体和过渡态存在时间又极短,实验室模拟反应存在一定的限制,导致计算给出相关的机制难以由实验证实。同时由于真实大气介质的化学组成与气象条件具有复杂性,理论计算尚无法完全考虑这些复杂因素;并且RGM的光解还原反应的计算涉及到大量激发态的计算,这对于计算资源有较高要求,因此理论模拟也存在一定的局限性。未来大气汞的氧化还原反应的机制研究工作可着重关注以下几点:
(1) 在不同环境条件下,因为大气中氧化剂或者还原剂的浓度不同、温度不同和压力不同,导致的具体环境中大气汞氧化还原反应机制存在哪些差异,这些差异对于大气汞的地球化学循环过程的影响有哪些。
(2) 目前采用量子化学方法对于大气汞在水相和非均相的研究还较少,未来可多开展大气汞在大气液相和颗粒物上的氧化还原反应机制的理论研究,这对于全面了解汞在大气中的迁移转化具有重要意义。
(3) 在理论研究的基础上,未来可采用更丰富和先进的实验方法与技术对大气汞的氧化还原机制进行验证,理论与实验方法相结合,获得更准确的大气汞氧化还原反应过程,来提高大气汞的传输和迁移转化模型的准确性。
大气汞氧化还原过程与机制的计算化学研究进展
Advances in computational chemistry for oxidation and reduction of mercury in the atmosphere
-
摘要: 大气中汞的氧化还原反应对于其全球生物地球化学循环起着极其重要的作用,它促进了汞在全球范围内的扩散。汞主要以气态元素汞的形态释放到大气中,并经历复杂的均相和非均相化学反应,被氧化为活性气态汞和颗粒态汞;同时,活性气态汞也可经过光致还原反应光解生成气态元素汞。计算化学是一种基于理论方法利用计算软件来对化学现象和本质进行解释和预测的方法,现已广泛用于大气汞氧化还原反应机制的研究。本文从计算化学角度出发,综述了大气中常见氧化剂包括O3、·OH、NO3和各种卤素自由基等对气态元素汞的氧化机制的理论研究,包括通过计算氧化产物的键解离能、键长和生成焓等热力学参数来研究反应产物的稳定性;通过计算反应的中间体和过渡态来阐述微观反应机制;通过计算反应速率常数来研究反应进行的快慢等。同时,本文还综述了最新报道的应用高精度量子化学计算方法研究活性气态汞还原为气态元素汞的反应机制,包括活性气态汞的紫外可见吸收光谱、势能面、光解反应速率以及光解反应的分支比等。了解大气汞的氧化还原反应的计算化学研究进展,对于深入了解汞在大气中的迁移转化具有至关重要的作用。Abstract: The redox reactions of atmospheric mercury play an important role in the biogeochemical cycle of mercury. Mercury is mainly released into the atmosphere as gaseous elemental mercury (GEM), which undergoes complex redox reactions to be oxidized to reactive gaseous mercury (RGM) and particulate bound mercury (PBM). Meanwhile, RGM also can be photolyzed to GEM. Computational chemistry is an approach covering any use of computing in the direct study of chemical problems, which is widely used in studying the mechanism of atmospheric mercury redox reactions. This review summarizes the theoretical studies of the oxidation mechanism of GEM by common atmospheric oxidants such as O3, ·OH, NO3 and halogen radicals, including bond dissociation energy, transition states, rate constant and so on. Meanwhile, this article also reviews the photodissociation mechanisms of RGM based on high-level quantum chemical methods, including the UV-Vis absorption spectra and cross-sections of RGM, two-dimensional potential energy surfaces, photodissociation yields and photolysis rates. Understanding the theoretical studies on the photochemistry of mercury in the atmosphere is helpful to understand the global atmospheric mercury distribution, transport, and lifetime.
-
-
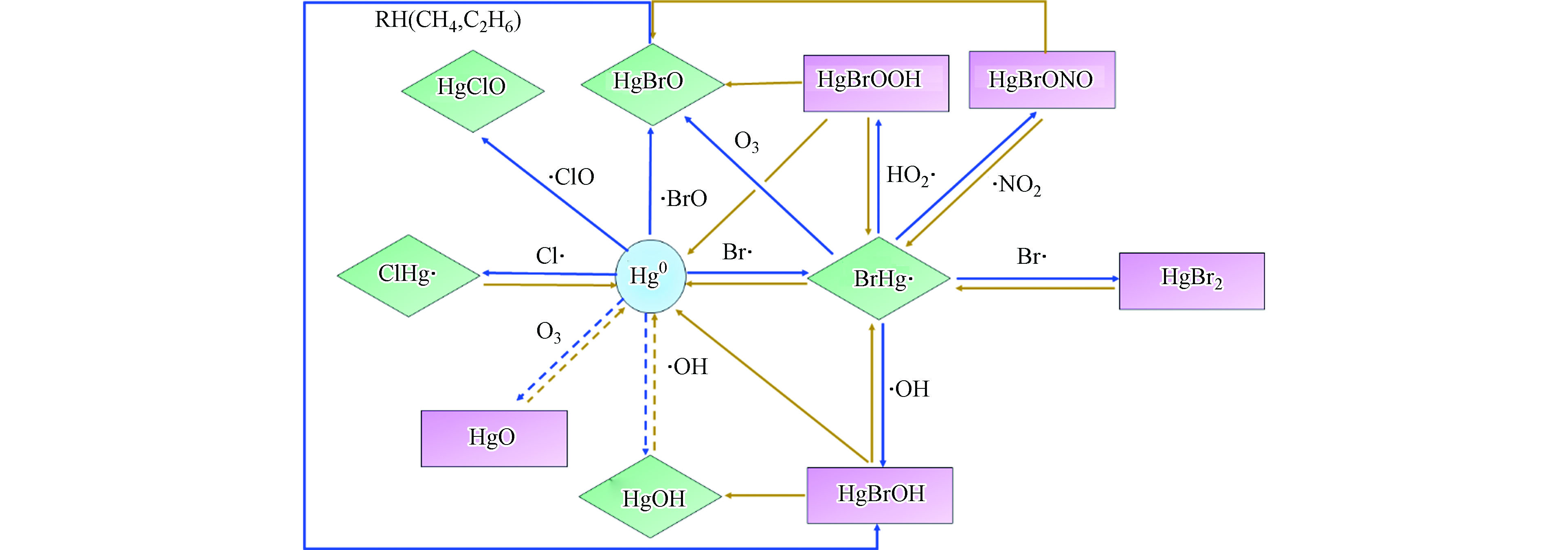
图 1 大气中不同形态汞的主要光化学转化机制。
Figure 1. Photochemical reactions of major mercury species in the atmosphere
表 1 BrHg—Y和ClHg—Y在0 K下的键解离能
Table 1. Bond dissociation energy of BrHg—Y and ClHg—Y at 0 K
BrHg—Y化合物
Compounds of BrHg—Y键离解能D0/(kJ·mol−1)
Bond dissociation energy
at 0 KClHg—Y化合物
Compounds of ClHg—Y键离解能D0/(kJ·mol−1)
Bond dissociation energy
at 0 KBrHg—Br 305.4[47], 303.8[44] ClHg—Cl 337.0[36] BrHg—NO2 138.9[48], 149.0[37], 142.7[40], 139.3[36] ClHg—NO2 153.4[36] anti-BrHg—ONO 156.1[48], 160.7[37], 151.9[40], 150.2[36] ClHg—ONO 165.0[36] syn-BrHg—ONO 177.0[48], 182.2[37], 177.4[40], 176.1[36] BrHg—OOH 177.4[37], 167.4[36] ClHg—OOH 183.1[36] BrHg—OBr 232.6[36], 223.8[37] ClHg—OBr 237.0[36] BrHg—OCl 220.6[36], 211.7[37] ClHg—OCl 225.1[36] BrHg—NO 49.2[37] ClHg—NO 54.4[36] BrHg—OO 27.9[36] ClHg—OO 33.0[36] BrHg—OI 54.8[37] BrHg—Cl 83.46[37] BrHg—I 70.06[37]  下载: 导出CSV
下载: 导出CSV
表 2 RGM的解离反应和解离速率常数
Table 2. Dissociation reaction and dissociation rate of RGM
解离反应
Dissociation reaction速率常数
Dissociation rate参考文献
ReferencesHgBr+M→Hg0 + Br +M 1.6×10−9 cm3·molecule−1·s−1 [36] HOHg+M → Hg0 + OH +M 1.22×10−9 cm3·molecule−1·s−1 [29] HgBr → Hg0 + Br 3×10−2 s−1 [15] HOHg → Hg0 + OH 1×10−2 s−1 [15] HgO → Hg0 + O 5.42×10−1 s−1 [65] HgBrO → Hg0 + Br + O 2.95×10−2 s−1 [9] HgBrO→ HgO+ Br 2.95×10−2 s−1 [9] HgBrOH →Hg0 + Br + OH 1.07×10−5 s−1 [65] HgBrOH →·HgOH + Br 1.07×10−5 s−1 [65] HgBrOH → HgBr + OH 1.07×10−5 s−1 [65] HgBrOH → HgBrO + H 1.07×10−5 s−1 [65] HgBrOOH→ Hg0 + Br + OOH 1.32×10−2 s−1 [65] HgBrOOH→ HgBrO + OH 1.32×10−2 s−1 [65] HgBrOOH→ HgBr + OOH 1.32×10−2 s−1 [65] HgBrOOH→ HgBrOH + O 1.32×10−2 s−1 [65] syn-HgBrONO → HgBrO+ NO 9.6×10−4 s−1 [65] syn-HgBrONO → HgBr + NO2 9.6×10−4 s−1 [65]
下载: 导出CSV
-
[1] 史建波, 阴永光, 江桂斌. 汞的分子转化与长距离传输[M]. 北京: 科学出版社, 2019. SHI J B, YIN Y G, JIANG G B. Molecular transformation and long-range transport of mercury [M]. Beijing: Science Press, 2019(in Chinese).
[2] SAIZ-LOPEZ A, TRAVNIKOV O, SONKE J E, et al. Photochemistry of oxidized Hg(I) and Hg(II) species suggests missing mercury oxidation in the troposphere [J]. PNAS, 2020, 117(49): 30949-30956. doi: 10.1073/pnas.1922486117 [3] 方莹莹, 王颖, 史建波, 等. 大气中活性气态汞的分析方法和赋存转化 [J]. 化学进展, 2021, 33(1): 151-161. FANG Y Y, WANG Y, SHI J B, et al. Analysis methods, occurrence, and transformation of reactive gaseous mercury in the atmosphere [J]. Progress in Chemistry, 2021, 33(1): 151-161(in Chinese).
[4] SCHROEDER W H, MUNTHE J. Atmospheric mercury—An overview [J]. Atmospheric Environment, 1998, 32(5): 809-822. doi: 10.1016/S1352-2310(97)00293-8 [5] LIN C J, PEHKONEN S O. The chemistry of atmospheric mercury: A review [J]. Atmospheric Environment, 1999, 33(13): 2067-2079. doi: 10.1016/S1352-2310(98)00387-2 [6] SHAO J J, SHI J B, DUO B, et al. Mercury in alpine fish from four rivers in the Tibetan Plateau [J]. Journal of Environmental Sciences, 2016, 39: 22-28. doi: 10.1016/j.jes.2015.09.009 [7] POISSANT L, ZHANG H H, CANÁRIO J, et al. Critical review of mercury fates and contamination in the arctic tundra ecosystem [J]. Science of the Total Environment, 2008, 400(1/2/3): 173-211. [8] KHIRI D, LOUIS F, ČERNUŠÁK I, et al. BrHgO• + CO: Analogue of OH + CO and reduction path for Hg(II) in the atmosphere [J]. ACS Earth and Space Chemistry, 2020, 4(10): 1777-1784. doi: 10.1021/acsearthspacechem.0c00171 [9] FRANCÉS-MONERRIS A, CARMONA-GARCÍA J, ACUÑA A U, et al. Photodissociation mechanisms of major mercury(II) species in the atmospheric chemical cycle of mercury [J]. Angewandte Chemie (International Ed. in English), 2020, 59(19): 7605-7610. doi: 10.1002/anie.201915656 [10] LU X, LIU Y R, JOHS A, et al. Anaerobic mercury methylation and demethylation by Geobacter bemidjiensis bem [J]. Environmental Science & Technology, 2016, 50(8): 4366-4373. [11] DENG L, WU F, DENG N S, et al. Photoreduction of mercury(II) in the presence of algae, Anabaena cylindrical [J]. Journal of Photochemistry and Photobiology B:Biology, 2008, 91(2/3): 117-124. [12] SCHROEDER W H, ANLAUF K G, BARRIE L A, et al. Arctic springtime depletion of mercury [J]. Nature, 1998, 394(6691): 331-332. doi: 10.1038/28530 [13] HOLMES C D, JACOB D J, CORBITT E S, et al. Global atmospheric model for mercury including oxidation by bromine atoms [J]. Atmospheric Chemistry and Physics, 2010, 10(24): 12037-12057. doi: 10.5194/acp-10-12037-2010 [14] WANG S Y, MCNAMARA S M, MOORE C W, et al. Direct detection of atmospheric atomic bromine leading to mercury and ozone depletion [J]. PNAS, 2019, 116(29): 14479-14484. doi: 10.1073/pnas.1900613116 [15] SAIZ-LOPEZ A, ACUÑA A U, TRABELSI T, et al. Gas-phase photolysis of Hg(I) radical species: A new atmospheric mercury reduction process [J]. Journal of the American Chemical Society, 2019, 141(22): 8698-8702. doi: 10.1021/jacs.9b02890 [16] ARIYA P A, KHALIZOV A, GIDAS A. Reactions of gaseous mercury with atomic and molecular halogens: kinetics, product studies, and atmospheric implications [J]. The Journal of Physical Chemistry A, 2002, 106(32): 7310-7320. doi: 10.1021/jp020719o [17] CALVERT J G, LINDBERG S E. Mechanisms of mercury removal by O3 and OH in the atmosphere [J]. Atmospheric Environment, 2005, 39(18): 3355-3367. doi: 10.1016/j.atmosenv.2005.01.055 [18] SHEPLER B C, PETERSON K A. Mercury monoxide: A systematic investigation of its ground electronic state [J]. The Journal of Physical Chemistry A, 2003, 107(11): 1783-1787. doi: 10.1021/jp027512f [19] FILATOV M, CREMER D. Revision of the dissociation energies of mercury chalcogenides: Unusual types of mercury bonding [J]. Chemphyschem, 2004, 5(10): 1547-1557. doi: 10.1002/cphc.200301207 [20] HALL B. The gas phase oxidation of elemental mercury by ozone [J]. Water, Air, and Soil Pollution, 1995, 80(1/2/3/4): 301-315. [21] TOSSELL J A. Calculation of the energetics for oxidation of gas-phase elemental Hg by Br and BrO [J]. The Journal of Physical Chemistry A, 2003, 107(39): 7804-7808. doi: 10.1021/jp030390m [22] PAL B, ARIYA P A. Studies of ozone initiated reactions of gaseous mercury: Kinetics, product studies, and atmospheric implications [J]. Physical Chemistry Chemical Physics, 2004, 6(3): 572. doi: 10.1039/b311150d [23] SUMNER A L, SPICER C W, SATOLA J, et al. Environmental chamber studies of mercury reactions in the atmosphere[M]//Dynamics of Mercury Pollution on Regional and Global Scales: . New York: Springer-Verlag, : 193-212. [24] SNIDER G, RAOFIE F, ARIYA P A. Effects of relative humidity and CO(g) on the O3-initiated oxidation reaction of Hg0(g): Kinetic & product studies [J]. Physical Chemistry Chemical Physics, 2008, 10(36): 5616. doi: 10.1039/b801226a [25] HYNES A J, DONOHOUE D L, GOODSITE M E, et al. Our current understanding of major chemical and physical processes affecting mercury dynamics in the atmosphere and at the air-water/terrestrial interfacesMercury Fate and Transport in the Global Atmosphere, 2009: 427-457. DOI:10.1007/978-0-387-93958-2_14. [26] SOMMAR J, GÅRDFELDT K, STRÖMBERG D, et al. A kinetic study of the gas-phase reaction between the hydroxyl radical and atomic mercury [J]. Atmospheric Environment, 2001, 35(17): 3049-3054. doi: 10.1016/S1352-2310(01)00108-X [27] GOODSITE M E, PLANE J M C, SKOV H. A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere [J]. Environmental Science & Technology, 2004, 38(6): 1772-1776. [28] EZARFI N, TOUIMI BENJELLOUN A, SABOR S, et al. Theoretical investigations of structural, thermal properties and stability of the group 12 metal M(XH) isomers in atmosphere: M = (Zn, Cd, Hg) and XH = (OH, SH) [J]. Theoretical Chemistry Accounts, 2019, 138(9): 1-14. [29] DIBBLE T S, TETU H L, JIAO Y G, et al. Modeling the OH-initiated oxidation of mercury in the global atmosphere without violating physical laws [J]. The Journal of Physical Chemistry. A, 2020, 124(2): 444-453. doi: 10.1021/acs.jpca.9b10121 [30] CREMER D, KRAKA E, FILATOV M. Bonding in mercury molecules described by the normalized elimination of the small component and coupled cluster theory [J]. Chemphyschem, 2008, 9(17): 2510-2521. doi: 10.1002/cphc.200800510 [31] BAUER D, D’OTTONE L, CAMPUZANO-JOST P, et al. Gas phase elemental mercury: A comparison of LIF detection techniques and study of the kinetics of reaction with the hydroxyl radical [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2003, 157(2/3): 247-256. [32] HOROWITZ H M, JACOB D J, ZHANG Y X, et al. A new mechanism for atmospheric mercury redox chemistry: Implications for the global mercury budget [J]. Atmospheric Chemistry and Physics, 2017, 17(10): 6353-6371. doi: 10.5194/acp-17-6353-2017 [33] WANG F, SAIZ-LOPEZ A, MAHAJAN A S, et al. Enhanced production of oxidised mercury over the tropical Pacific Ocean: A key missing oxidation pathway [J]. Atmospheric Chemistry and Physics, 2014, 14(3): 1323-1335. doi: 10.5194/acp-14-1323-2014 [34] SHEPLER B C, BALABANOV N B, PETERSON K A. Ab initio thermochemistry involving heavy atoms: an investigation of the reactions Hg + IX (X = I, br, cl, O) [J]. The Journal of Physical Chemistry A, 2005, 109(45): 10363-10372. doi: 10.1021/jp0541617 [35] TELLINGHUISEN J, ASHMORE J G. Mixed representations for diatomic spectroscopic data: Application to HgBr [J]. Chemical Physics Letters, 1983, 102(1): 10-16. doi: 10.1016/0009-2614(83)80647-2 [36] DIBBLE T S, ZELIE M J, MAO H. Thermodynamics of reactions of ClHg and BrHg radicals with atmospherically abundant free radicals [J]. Atmospheric Chemistry and Physics, 2012, 12(21): 10271-10279. doi: 10.5194/acp-12-10271-2012 [37] JIAO Y G, DIBBLE T S. Quality structures, vibrational frequencies, and thermochemistry of the products of reaction of BrHg• with NO2, HO2, ClO, BrO, and IO [J]. The Journal of Physical Chemistry A, 2015, 119(42): 10502-10510. doi: 10.1021/acs.jpca.5b04889 [38] DIBBLE T S, SCHWID A C. Thermodynamics limits the reactivity of BrHg radical with volatile organic compounds [J]. Chemical Physics Letters, 2016, 659: 289-294. doi: 10.1016/j.cplett.2016.07.065 [39] TOYOTA K, DASTOOR A P, RYZHKOV A. Air–snowpack exchange of bromine, ozone and mercury in the springtime Arctic simulated by the 1-D model PHANTAS – Part 2: Mercury and its speciation [J]. Atmospheric Chemistry and Physics, 2014, 14(8): 4135-4167. doi: 10.5194/acp-14-4135-2014 [40] JIAO Y G, DIBBLE T S. First kinetic study of the atmospherically important reactions BrHg˙ + NO2 and BrHg˙ + HOO [J]. Physical Chemistry Chemical Physics, 2017, 19(3): 1826-1838. doi: 10.1039/C6CP06276H [41] DONOHOUE D L, BAUER D, COSSAIRT B, et al. Temperature and pressure dependent rate coefficients for the reaction of Hg with Br and the reaction of Br with br: A pulsed laser photolysis-pulsed laser induced fluorescence study [J]. The Journal of Physical Chemistry A, 2006, 110(21): 6623-6632. doi: 10.1021/jp054688j [42] SUN G Y, SOMMAR J, FENG X B, et al. Mass-dependent and -independent fractionation of mercury isotope during gas-phase oxidation of elemental mercury vapor by atomic Cl and Br [J]. Environmental Science & Technology, 2016, 50(17): 9232-9241. [43] KHALIZOV A F, VISWANATHAN B, LARREGARAY P, et al. A theoretical study on the reactions of Hg with halogens: atmospheric implications [J]. The Journal of Physical Chemistry A, 2003, 107(33): 6360-6365. doi: 10.1021/jp0350722 [44] SHEPLER B C, BALABANOV N B, PETERSON K A. Hg+Br→HgBr recombination and collision-induced dissociation dynamics [J]. The Journal of Chemical Physics, 2007, 127(16): 164304. doi: 10.1063/1.2777142 [45] RAOFIE F, ARIYA P A. Product study of the gas-phase BrO-initiated oxidation of Hg0: Evidence for stable Hg1+ compounds [J]. Environmental Science & Technology, 2004, 38(16): 4319-4326. [46] TAS E, OBRIST D, PELEG M, et al. Measurement-based modelling of bromine-induced oxidation of mercury above the Dead Sea [J]. Atmospheric Chemistry and Physics, 2012, 12(5): 2429-2440. doi: 10.5194/acp-12-2429-2012 [47] BALABANOV N B, PETERSON K A. Mercury and reactive halogens: The thermochemistry of Hg + {Cl2, Br2, BrCl, ClO, and BrO} [J]. The Journal of Physical Chemistry A, 2003, 107(38): 7465-7470. doi: 10.1021/jp035547p [48] DIBBLE T S, ZELIE M J, JIAO Y G. Quantum chemistry guide to PTRMS studies of as-yet undetected products of the bromine-atom initiated oxidation of gaseous elemental mercury [J]. The Journal of Physical Chemistry A, 2014, 118(36): 7847-7854. doi: 10.1021/jp5041426 [49] TOKOS J J S, HALL B, CALHOUN J A, et al. Homogeneous gas-phase reaction of Hg° with H2O2,CH3I, AND (CH3)2S: Implications for atmospheric Hg cycling [J]. Atmospheric Environment, 1998, 32(5): 823-827. doi: 10.1016/S1352-2310(97)00171-4 [50] SOMMAR J, HALLQUIST M, LJUNGSTRÖM E, et al. On the gas phase reactions between volatile biogenic mercury species and the nitrate radical [J]. Journal of Atmospheric Chemistry, 1997, 27(3): 233-247. doi: 10.1023/A:1005873712847 [51] PELEG M, TAS E, OBRIST D, et al. Observational evidence for involvement of nitrate radicals in nighttime oxidation of mercury [J]. Environmental Science & Technology, 2015, 49(24): 14008-14018. [52] RUTTER A P, SHAKYA K M, LEHR R, et al. Oxidation of gaseous elemental mercury in the presence of secondary organic aerosols [J]. Atmospheric Environment, 2012, 59: 86-92. doi: 10.1016/j.atmosenv.2012.05.009 [53] KIM P R, HAN Y J, HOLSEN T M, et al. Atmospheric particulate mercury: Concentrations and size distributions [J]. Atmospheric Environment, 2012, 61: 94-102. doi: 10.1016/j.atmosenv.2012.07.014 [54] CHEN J B, HINTELMANN H, FENG X B, et al. Unusual fractionation of both odd and even mercury isotopes in precipitation from Peterborough, ON, Canada [J]. Geochimica et Cosmochimica Acta, 2012, 90: 33-46. doi: 10.1016/j.gca.2012.05.005 [55] KURIEN U, HU Z Z, LEE H, et al. Radiation enhanced uptake of Hg0(g) on iron (oxyhydr)oxide nanoparticles [J]. RSC Advances, 2017, 7(71): 45010-45021. doi: 10.1039/C7RA07401H [56] PIRRONE N, CINNIRELLA S, FENG X, et al. Global mercury emissions to the atmosphere from anthropogenic and natural sources [J]. Atmospheric Chemistry and Physics, 2010, 10(13): 5951-5964. doi: 10.5194/acp-10-5951-2010 [57] HU Y, CHENG H F. Control of mercury emissions from stationary coal combustion sources in China: Current status and recommendations [J]. Environmental Pollution, 2016, 218: 1209-1221. doi: 10.1016/j.envpol.2016.08.077 [58] GALBREATH K C, ZYGARLICKE C J. Mercury transformations in coal combustion flue gas [J]. Fuel Processing Technology, 2000, 65/66: 289-310. doi: 10.1016/S0378-3820(99)00102-2 [59] PAVLISH J H, SONDREAL E A, MANN M D, et al. Status review of mercury control options for coal-fired power plants [J]. Fuel Processing Technology, 2003, 82(2/3): 89-165. [60] HALL B, SCHAGER P, LINDQVIST O. Chemical reactions of mercury in combustion flue gases [J]. Water Air & Soil Pollution, 1991, 56(1): 3-14. [61] NIKSA S, NAIK C V, BERRY M S, et al. Interpreting enhanced Hg oxidation with Br addition at Plant Miller [J]. Fuel Processing Technology, 2009, 90(11): 1372-1377. doi: 10.1016/j.fuproc.2009.05.022 [62] NORTON G A, YANG H Q, BROWN R C, et al. Heterogeneous oxidation of mercury in simulated post combustion conditions [J]. Fuel, 2003, 82(2): 107-116. doi: 10.1016/S0016-2361(02)00254-5 [63] CZAPLICKA M, PYTA H. Transformations of mercury in processes of solid fuel combustion – review [J]. Archives of Environmental Protection, 2017, 43(4): 82-93. doi: 10.1515/aep-2017-0041 [64] YANG Y J, LIU J, WANG Z, et al. Homogeneous and heterogeneous reaction mechanisms and kinetics of mercury oxidation in coal-fired flue gas with bromine addition [J]. Proceedings of the Combustion Institute, 2017, 36(3): 4039-4049. doi: 10.1016/j.proci.2016.08.068 [65] SAIZ-LOPEZ A, SITKIEWICZ S P, ROCA-SANJUÁN D, et al. Photoreduction of gaseous oxidized mercury changes global atmospheric mercury speciation, transport and deposition [J]. Nature Communications, 2018, 9: 4796. doi: 10.1038/s41467-018-07075-3 [66] de FOY B, TONG Y D, YIN X F, et al. First field-based atmospheric observation of the reduction of reactive mercury driven by sunlight [J]. Atmospheric Environment, 2016, 134: 27-39. doi: 10.1016/j.atmosenv.2016.03.028 [67] HUANG Q, CHEN J B, HUANG W L, et al. Diel variation in mercury stable isotope ratios records photoreduction of PM2.5-bound mercury [J]. Atmospheric Chemistry and Physics, 2019, 19(1): 315-325. doi: 10.5194/acp-19-315-2019 [68] LAM K T, WILHELMSEN C J, SCHWID A C, et al. Computational study on the photolysis of BrHgONO and the reactions of BrHgO• with CH4, C2H6, NO, and NO2: Implications for formation of Hg(II) compounds in the atmosphere [J]. The Journal of Physical Chemistry A, 2019, 123(8): 1637-1647. doi: 10.1021/acs.jpca.8b11216 [69] DUZY C, HYMAN H A. Radiative lifetimes for the B→X transition in HgCl, HgBr, and HgI [J]. Chemical Physics Letters, 1977, 52(2): 345-348. doi: 10.1016/0009-2614(77)80556-3 [70] WADT W R. The electronic structure of HgCl and HgBr [J]. Applied Physics Letters, 1979, 34(10): 658-660. doi: 10.1063/1.90627 [71] JULIENNE P S, KONOWALOW D D, KRAUSS M, et al. Photodissociation of HgCl [J]. Applied Physics Letters, 1980, 36(2): 132-134. doi: 10.1063/1.91396 [72] KRAUSS M, STEVENS W J. Photodissociation of HgBr, XΣ1/2 [J]. Applied Physics Letters, 1981, 39(9): 686-688. doi: 10.1063/1.92869 [73] ARIYA P A, AMYOT M, DASTOOR A, et al. Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: A review and future directions [J]. Chemical Reviews, 2015, 115(10): 3760-3802. doi: 10.1021/cr500667e [74] YUE L, ZHOU S D, SUN X Y, et al. Direct room-temperature conversion of methane into protonated formaldehyde: The gas-phase chemistry of mercury among the zinc triad oxide cations [J]. Angewandte Chemie (International Ed. in English), 2018, 57(12): 3251-3255. doi: 10.1002/anie.201712405 [75] KAUPP M, von SCHNERING H G. Gaseous mercury(IV) fluoride, HgF4: An ab initio study [J]. Angewandte Chemie International Edition in English, 1993, 32(6): 861-863. doi: 10.1002/anie.199308611 [76] PONGPRUEKSA P, LIN C J, LINDBERG S E, et al. Scientific uncertainties in atmospheric mercury models III: Boundary and initial conditions, model grid resolution, and Hg(II) reduction mechanism [J]. Atmospheric Environment, 2008, 42(8): 1828-1845. doi: 10.1016/j.atmosenv.2007.11.020 [77] SEIGNEUR C, VIJAYARAGHAVAN K, LOHMAN K. Atmospheric mercury chemistry: Sensitivity of global model simulations to chemical reactions [J]. Journal of Geophysical Research Atmospheres, 2006, 111(D22): D22306. doi: 10.1029/2005JD006780 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 7221
- HTML全文浏览数: 7221
- PDF下载数: 142
- 施引文献: 0
